第4章 コマンドラインの使い方
分子構造のロード
loadコマンドは様々なファイルフォーマットを読み込むことができます。読み込もうとするファイルの拡張子に対応して自動的に読み込み形式を判定してくれます。例えばPDBファイルを正しく読み込む場合は.pdbである必要があり、MOLファイルであれば.mol、Macromodelファイルは.mmod、XPLOR mapファイルは.xplor、CCP4 mapファイルは.ccp4、Raster3Dインプットファイル(Molscript output)は.r3d、PyMOLセッションファイルは.pse、pickleになったChemPyモデル.pklなどは直接読み込むことができます。
以下の入力拡張子は右側のファイルタイプとして認識されます。
| 入力拡張子 | 認識されるファイルタイプ |
|---|---|
ent, p5m | pdb |
mmd, out, dat | mmod |
map, mrc | ccp4 |
cc2 | cc1 |
sd | sdf |
rst7 | rst |
o, dsn6, omap | brix |
ph4 | moe |
spi | spider |
pym, pyc | py |
p1m, pim | pml |
xml | pdbml |
loadコマンドを使うときにobject引数が指定された場合には、そのファイルは同名のオブジェクト上にファイルを読み込みます。指定しない場合、拡張子部分を除いたファイル名と同じ名前のオブジェクトが生成されます。
使い方
load filename [,object [,state [,format [,finish [,discrete [,multiplex ]]]]]]
PYMOL API
cmd.load( filename [,object [,state [,format [,finish [,discrete [,multiplex ]]]]]] )
引数
filename: (string) ロードしたいファイルのファイルパス、またはURLで指定することもできます。object: (string) 構造ファイルのオブジェクト名です。デフォルト名はロードするファイルのプレフィクスです。state: (integer) 読み込む構造ファイルをオブジェクトに保存する時、指定した数字のstateの上に上書きする形で読み込みます。例えば、MDのトラジェクトリファイルをオブジェクトの上に読み込む場合は、state=1を指定すると初期座標を上書きして(削除して)表示できるようになります。0を指定した場合は最後のstateの後に追加する形で読み込みます。(default:0)format: (string) ファイルのフォーマット形式を指定できます(see notes)。デフォルトはファイルの拡張子です。finish: (integer)discrete: (integer) MDトラジェクトリやNMR構造ファイルをロードしようとするようなマルチモデル構造に対しての場合、0に設定すると同じ原子セットを持つモデルであることを宣言し、メモリを節約することができます。1に設定すると強制的に各モデルについて別々の原子セットモデルを生成することができます。デフォルトは-1でファイルタイプに依存する設定になっています。(see discrete objects)quiet: (integer) デフォルトは1です。- multiplex : integer Load a multi-model file as separate objects instead of states (see also split_states)
- zoom : integer {default: -1 = use auto_zoom setting}
- partial : integer For session files (.pse). partial=0: discard current session. partial=1: merge with current session (will not load global settings, selections, movie, camera). partial=2: like 1, but also load camera view {default: 0}
- mimic : integer For .mae files, match style from file as close as possible, uses atom-level settings (like cartoon_color) {default: 1}
- object_props : string = Space delimited list of property names (or * wildcard) to load from .sdf or .mae files {default: use load_object_props_default setting} Incentive PyMOL 1.6+
- atom_props : string = Space delimited list of property names (or * wildcard) to load from .mae files {default: use load_atom_props_default setting} Incentive PyMOL 1.6+
保存
分子構造の表示形式のON/OFF
オブジェクトの重ね合わせ
生体分子、特にタンパク質には、構成されるアミノ酸配列が異なるものの概してよく似た構造を取っているものがよくあります。新種のタンパク質について構造解析を行い、既存のタンパク質と構造・機能上よく似たタンパク質が存在した場合には、その構造を比べてみて類似点・相違点を述べるというのが構造解析論文のディスカッションでよく見られる光景です。
重ね合わせのためのコマンドはalignまたはsuperが用意されています。この2つのコマンドはともに構造の重ね合わせをすることができますが、配列相同性が高い場合はalignコマンドを、低い場合はsuperを利用することがそれぞれ推奨されています。アルゴリズム的に見ると、alignは構成アミノ酸配列を考慮するのに対し、superは配列を考慮せずに構造ベースで重ね合わせようとします。
alignコマンド
使い方
align mobile, target [, cutoff [, cycles
[, gap [, extend [, max_gap [, object
[, matrix [, mobile_state [, target_state
[, quiet [, max_skip [, transform [, reset ]]]]]]]]]]]]]
mobile= string: 移動するべきオブジェクトのatom selectiontarget= string: 重ね合わせる先のオブジェクトのatom selectioncutoff= float: RMS(root mean square :構造のズレの数値指標)を基準に、この値を超えると外れ値とみなして重ね合わせに考慮に入れなくする。 デフォルトは2.0。cycles= int: 重ね合わせ試行の繰り返し数。この回数分だけ、重ね合わせる→大きくずれている箇所を検出し、そこを考慮しないで再度重ね合わせ→……を行う。デフォルトは5回。gap,extend,max_gap: 配列アライメント上でのペナルティパラメータ。object= string: 重ね合わせ結果をアライメントオブジェクトとして出力するときのオブジェクト名を指定する。デフォルトでは重ね合わせオブジェクトを生成しない。matrix= string: 配列アライメントでの置換行列を指定する。デフォルトはBLOSUM62置換行列。mobile_state= int:mobileで指定したオブジェクトに複数のstateが存在する場合、どのstateを使ってアライメントするかを指定できる。デフォルトは0(全state)。target_state= int:targetで指定したオブジェクトに複数のstateが存在する場合、どのstateを使ってアライメントするかを指定できる。デフォルトは0quiet=0の場合、詳細なアウトプットを表示する。1の場合は表示しない。デフォルトはコマンドラインから利用した場合0で、APIから呼び出した場合は1。max_skip= ?transform=1ならばmobileオブジェクトを移動させる。0ならば移動させない。reset= ?
superコマンド
使い方
ほぼalignコマンドと同じですが、さらにオプションが追加されています。
super mobile, target [, cutoff [, cycles [, gap [, extend [, max_gap [, object [, matrix [, mobile_state [, target_state [, quiet [, max_skip [, transform [, reset [, seq [, radius [, scale [, base [, coord [, expect [, window [, ante ]]]]]]]]]]]]]]]]]]]]]
reset= ?seq= ?radius= ?scale= ?base= ?coord= ?expect= ?window= ?ante= ?
実例
ここではPDB IDの1alkと3q3qの2つのアルカリホスファターゼを例にとって構造の重ね合わせをしてみましょう。この2つはアルカリホスファターゼでありながら、全体の構造は大きく異なっています。また1alkの方は2量体であるのに対して3q3qの方は単量体で表示されていることに注意してください。
fetch 1alk
fetch 3q3q
alignを使って構造を重ね合わせてみるとこのような形になります。
align 1alk, 3q3q, object=objalign
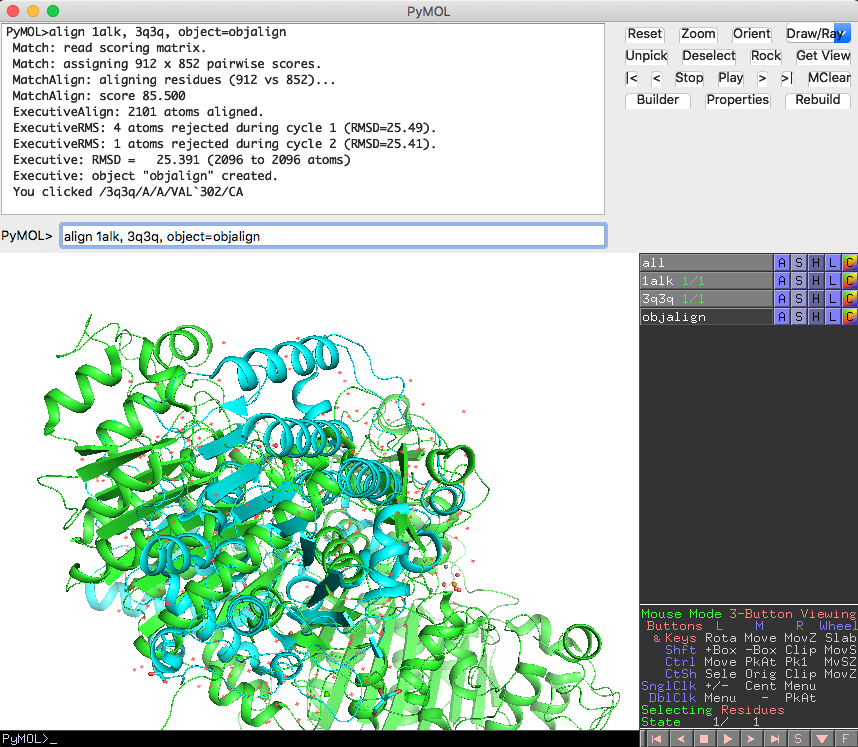
External GUIの出力結果を見ると、
Executive: RMSD = 25.391 (2096 to 2096 atoms)
となっており、構造のずれを表すRMSD値は25.3とかなり大きな値になっています。
一方で、superコマンドを使ってみると
super 1alk, 3q3q, object=objsuper
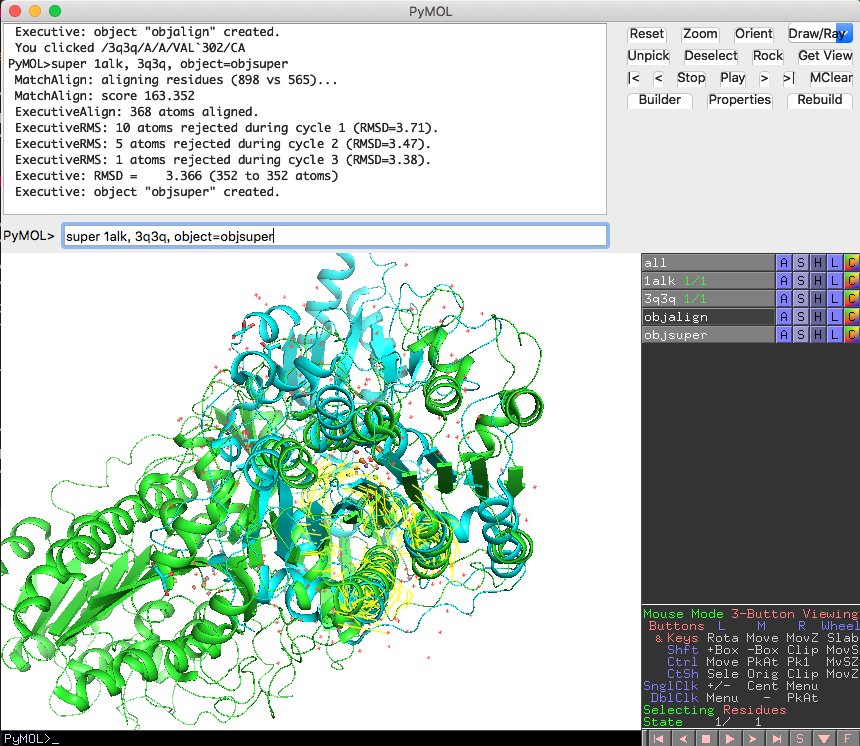
Executive: RMSD = 3.366 (352 to 352 atoms)
となっており、重ね合わせに利用した原子が352 atomsであり、その範囲でのRMSD値が3.366となりました。
また、objalignとobjsuperというアライメントオブジェクトが生成されています。このオブジェクトは2つの構造の重ね合わせのときに使った対応部分を黄色い線で表してくれています。superを行った場合は、1alk, 3q3qに共通しているαβα-sandwich構造の部分だけを自動的にサーチして、その部分だけを利用した重ね合わせを実行してくれています。