オブジェクトパネル
PyMOL画面の右側には、ロードしている分子構造について様々な操作を行えるパネルが表示されています。
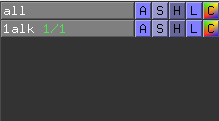
画像の例ではallと1alkが表示されています。また、画面上の分子構造をクリックし選択すると、(sele)という表示も追加されます。このパネルに表示されている文字のことをオブジェクトと呼びます。その右についているA, S, H, L, Cという文字のボタンには、対応するオブジェクトについての表示設定変更・編集を行うためのメニューがたくさん格納されています。それぞれの文字が表す大まかな意味は以下の通り。
- Action(A)
- 視点の移動や分子についての編集、一括設定変更、構造のアラインや名前変更など、オブジェクト全体に関わる様々な設定を行うためのメニューが格納されています。
- Show (S)
- 分子構造の表示形式をONにするためのメニューが格納されています。
- Hide (H)
- 分子構造の表示形式をOFFにするためのメニューが格納されています。
- Label (L)
- 分子構造のすぐそばに表示する文字ラベルを追加するためのメニューが格納されています。
- Color (C)
- 分子構造の色変更を行うためのメニューが格納されています。
分子構造の表示形式のON/OFF (Show and Hide)
タンパク質の構造情報をロードしたとき、デフォルト設定では、20種類のアミノ酸についてはCartoon表示形式と呼ばれる漫画風な表現形式になっています。このCartoon表示形式では、αヘリックスを形成している部分については大きな螺旋で描かれ、βストランド(βシート)を形成している部分については、進行方向に向かって矢印が伸びる表現で描かれています。
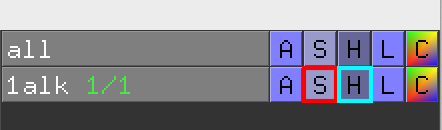
タンパク質を構成するアミノ酸を1つ1つ強調して見てみたい場合には、Cartoon, Line, Stick, Ribbon, Sphere, Dot, Surface, Mesh 表示などの、他の表示形式をONにするという操作を行います。この表示形式の設定は、各オブジェクト構造について個別に行います。例えば、先程ロードした1alkのタンパク質オブジェクトについて他の表示形式をONにしたい場合は、図のように、1alkのSマーク部分(図の赤四角部分のボタン) をクリックします。反対に、OFFにしたい場合はHマーク部分(図の青四角部分のボタン) を押し、各表示形式を選択することでOFFになります。
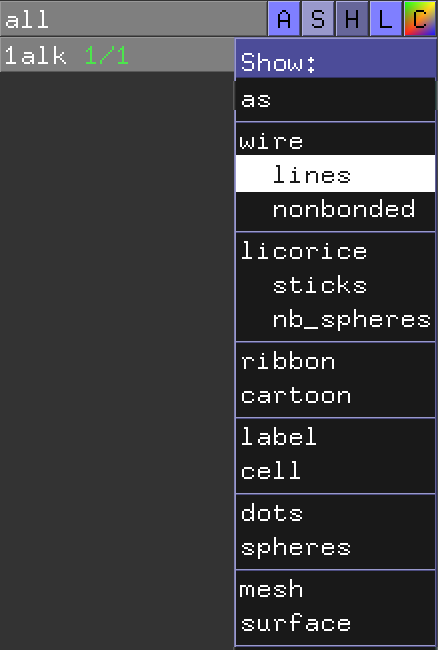
各表示形式のサンプルを以下に示します。
-
Cartoon表示
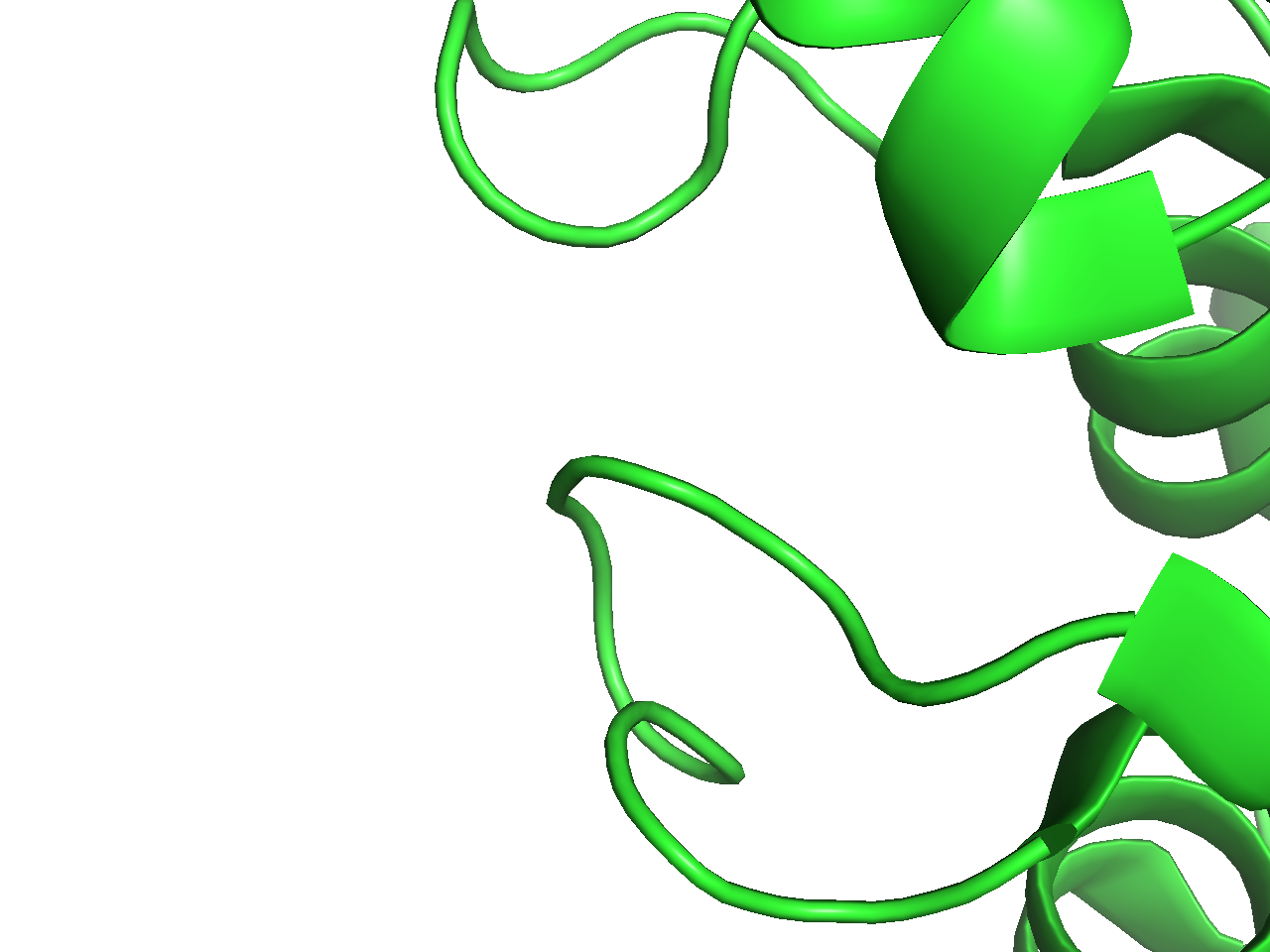
- タンパク質やDNA・RNAを表示するときにおすすめです。タンパク質の場合はαヘリックスやβシートが先述のように螺旋や矢印で示されます。DNA/RNAが存在する場合には、各塩基の概形や5’末端→3’末端のつながりが強調して表示されます。
-
Line表示
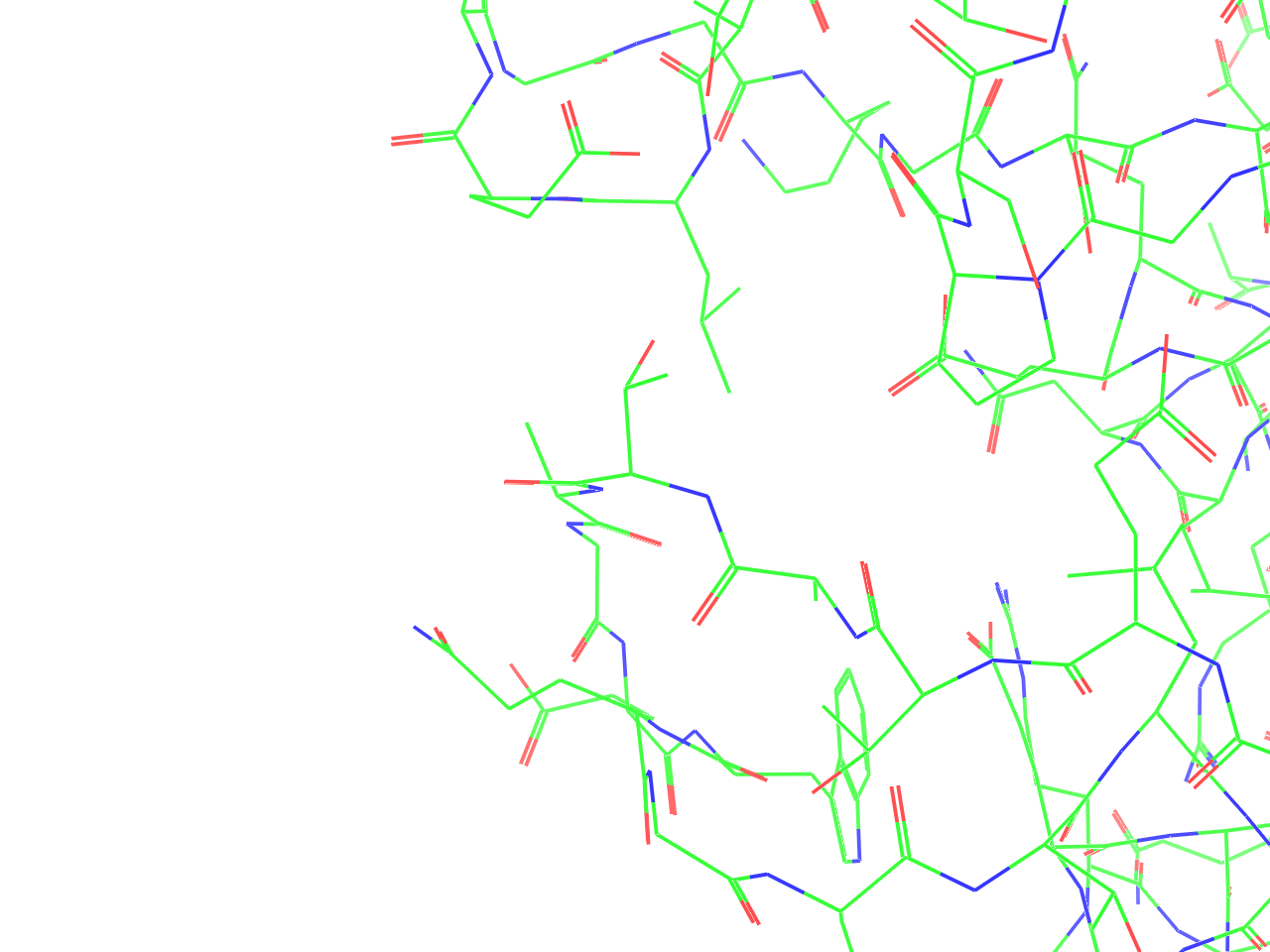
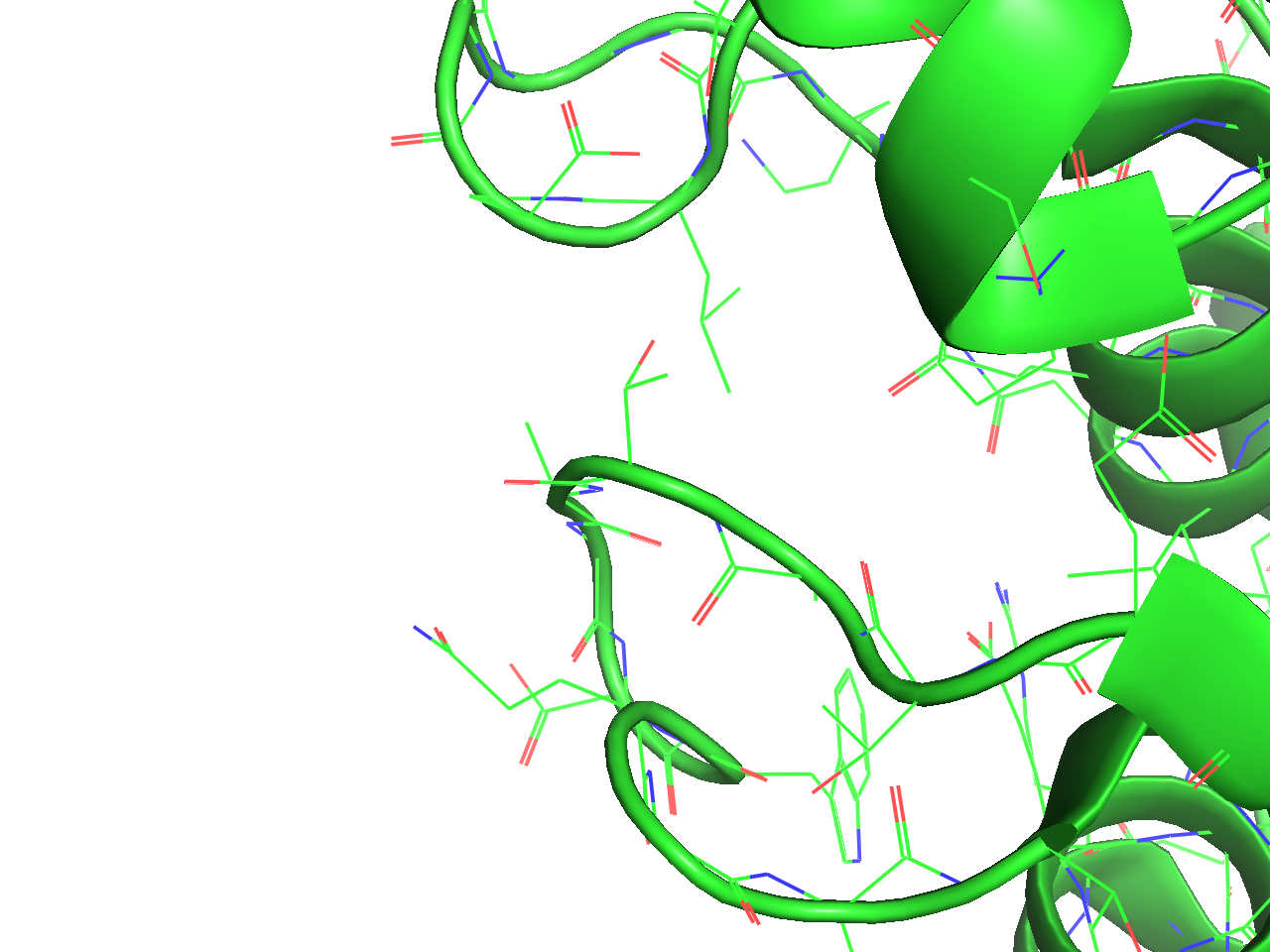
- 各原子間の結合が細い線で表示されます。上の図では、左にLine表示だけをONにしたものを、右にCartoon表示形式をLine表示とともにONにした場合を右図に示しています(以降、見やすくするためにCartoon表示をONにした上で、他の表示形式を載せます)。
- 各原子は構造情報ファイルの座標のATOMまたはHETATMレコードに書かれている座標情報(\( x, y, z \)の3次元ベクトル)をもとにプロットされ、各原子座標の距離が一定以内にあれば、自動的にその近い2点が直線で結ばれて表示される仕様となっていることに気をつけてください。
-
Stick表示
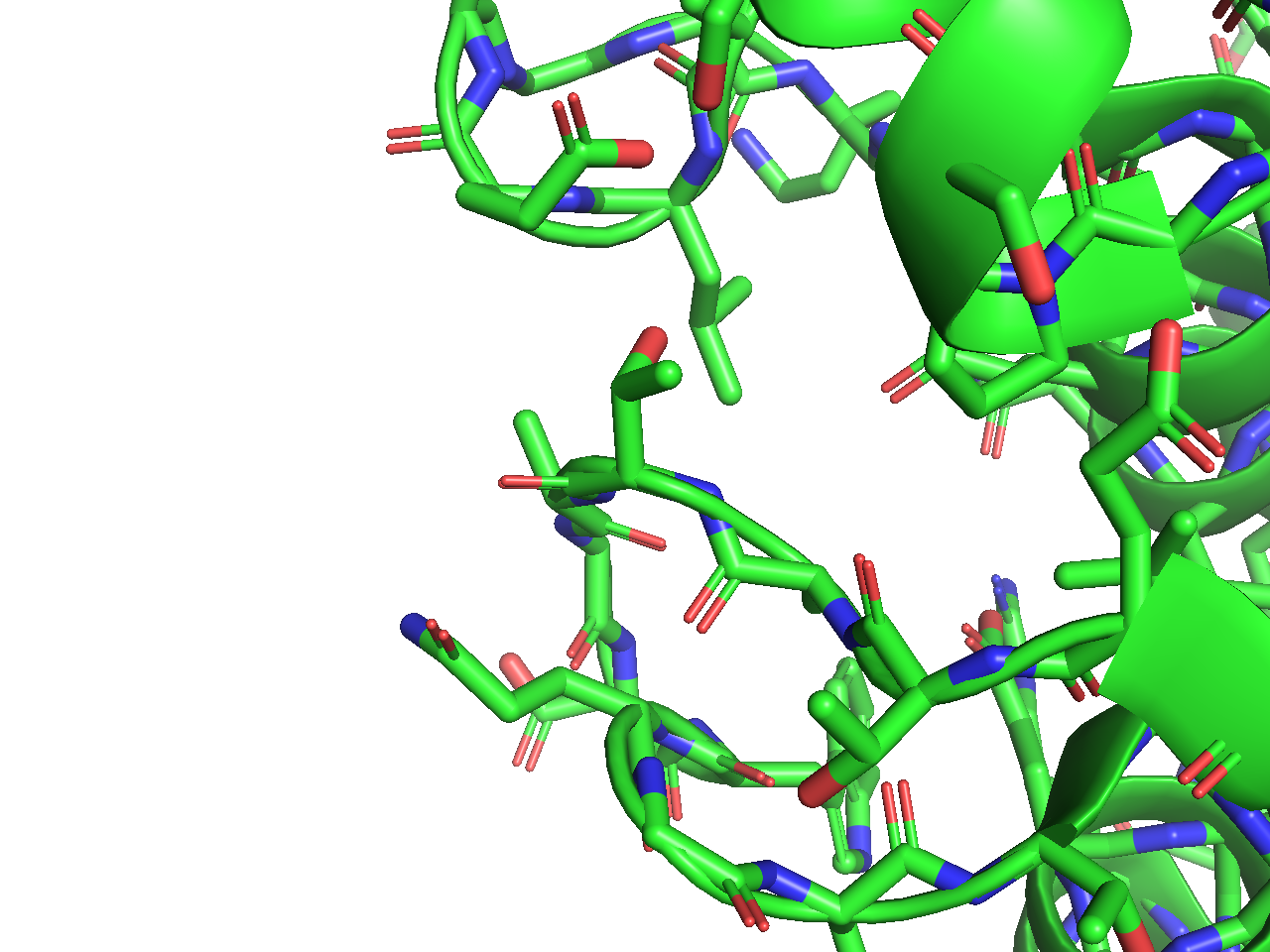
- 各原子間の結合がLine表示よりも太いStickで表示されます。Stickは円筒状に表現されます。
-
Ribbon表示

- タンパク質または核酸の鎖部分のみを単純な線で表現する形式です。
-
Sphere表示
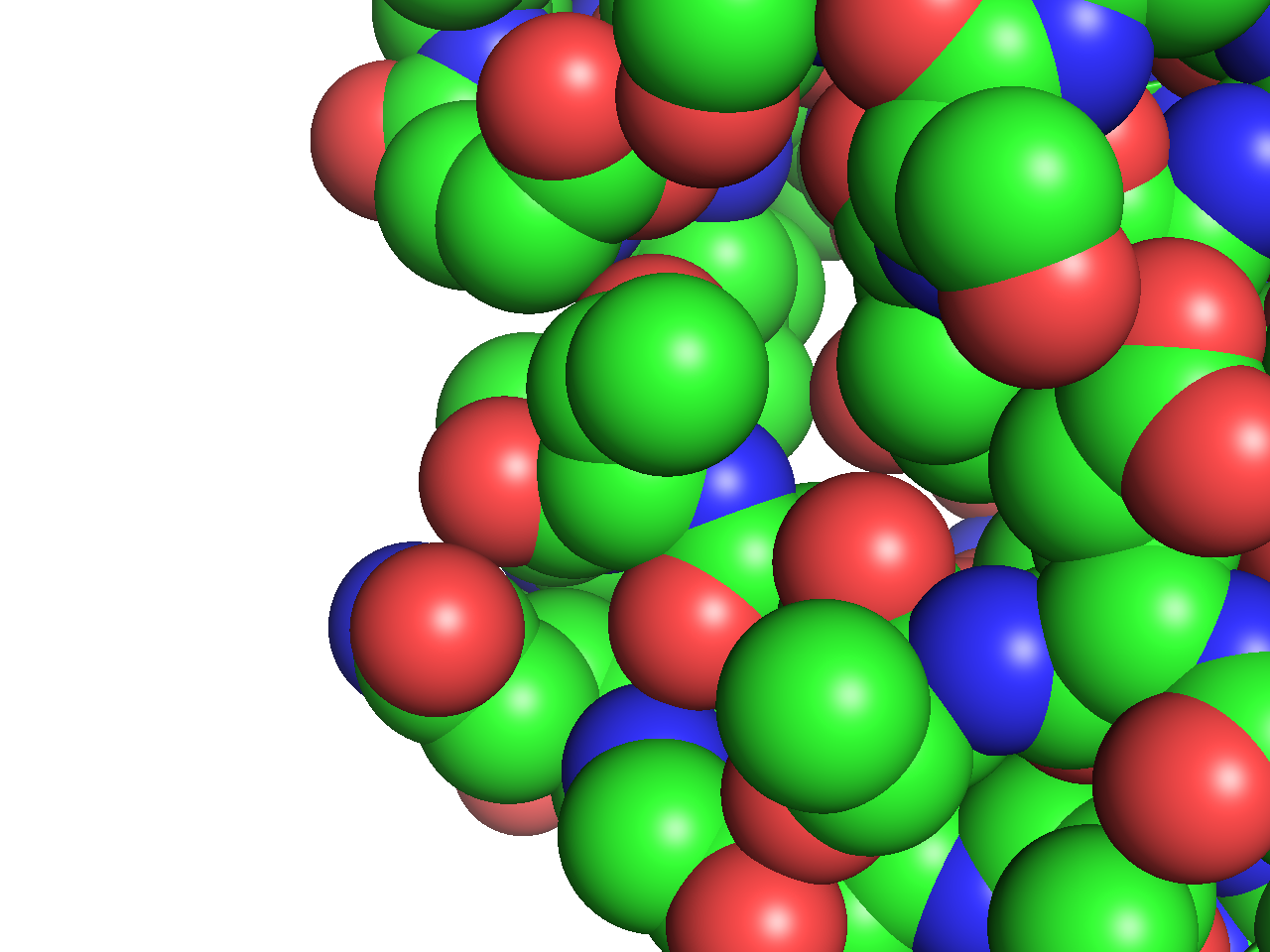
- 各原子を大きな球体で表現します。Line, Stick表示と異なり、各原子間は直線で結ばれません。
-
Dot表示
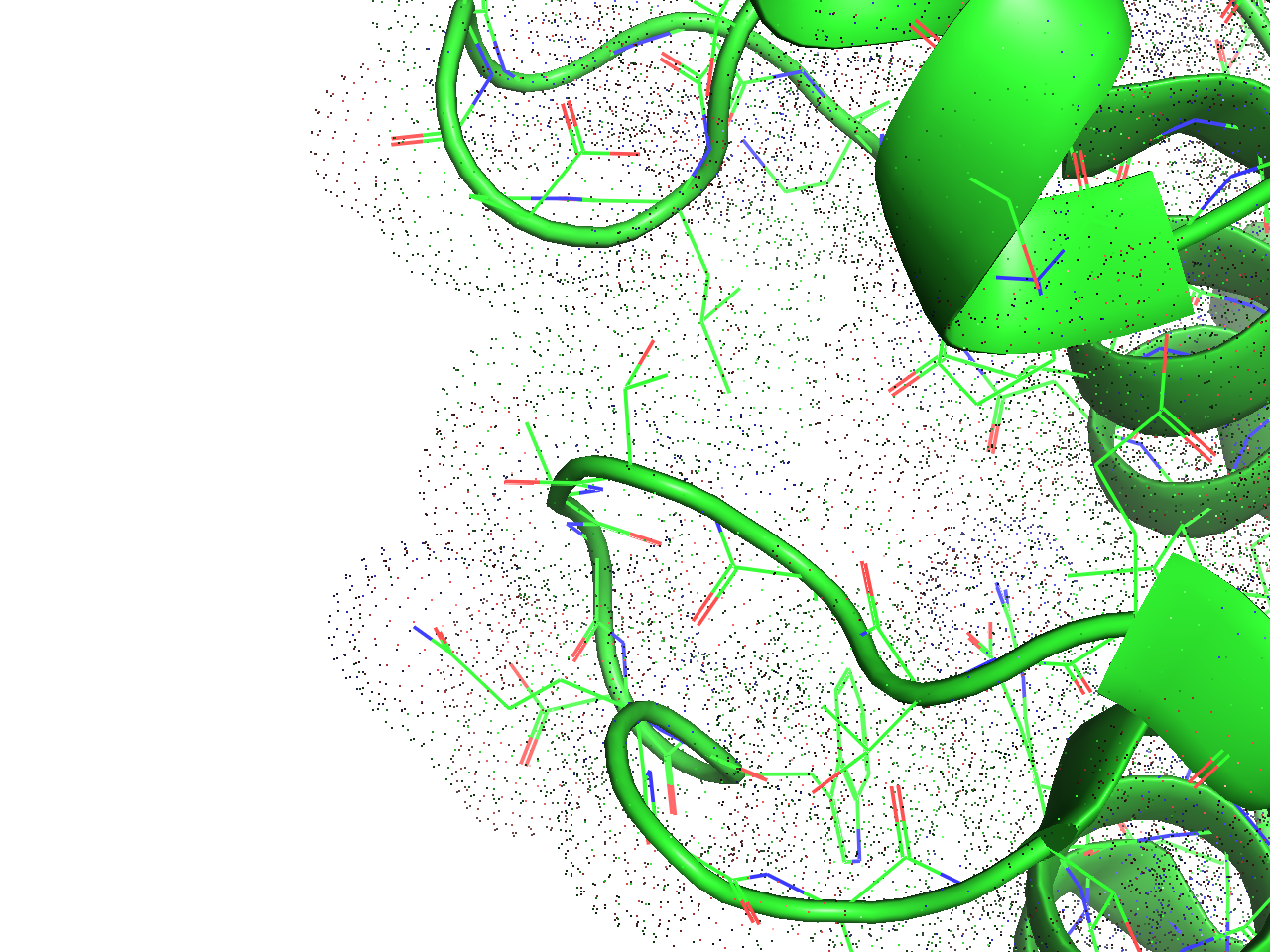
- Sphere表示の表面を点で表現したような形式です。
-
Surface表示

- 各原子の表面を滑らかに描画する形式です。分子の大きさによっては、レンダリングに少し時間がかかります。
-
Mesh表示
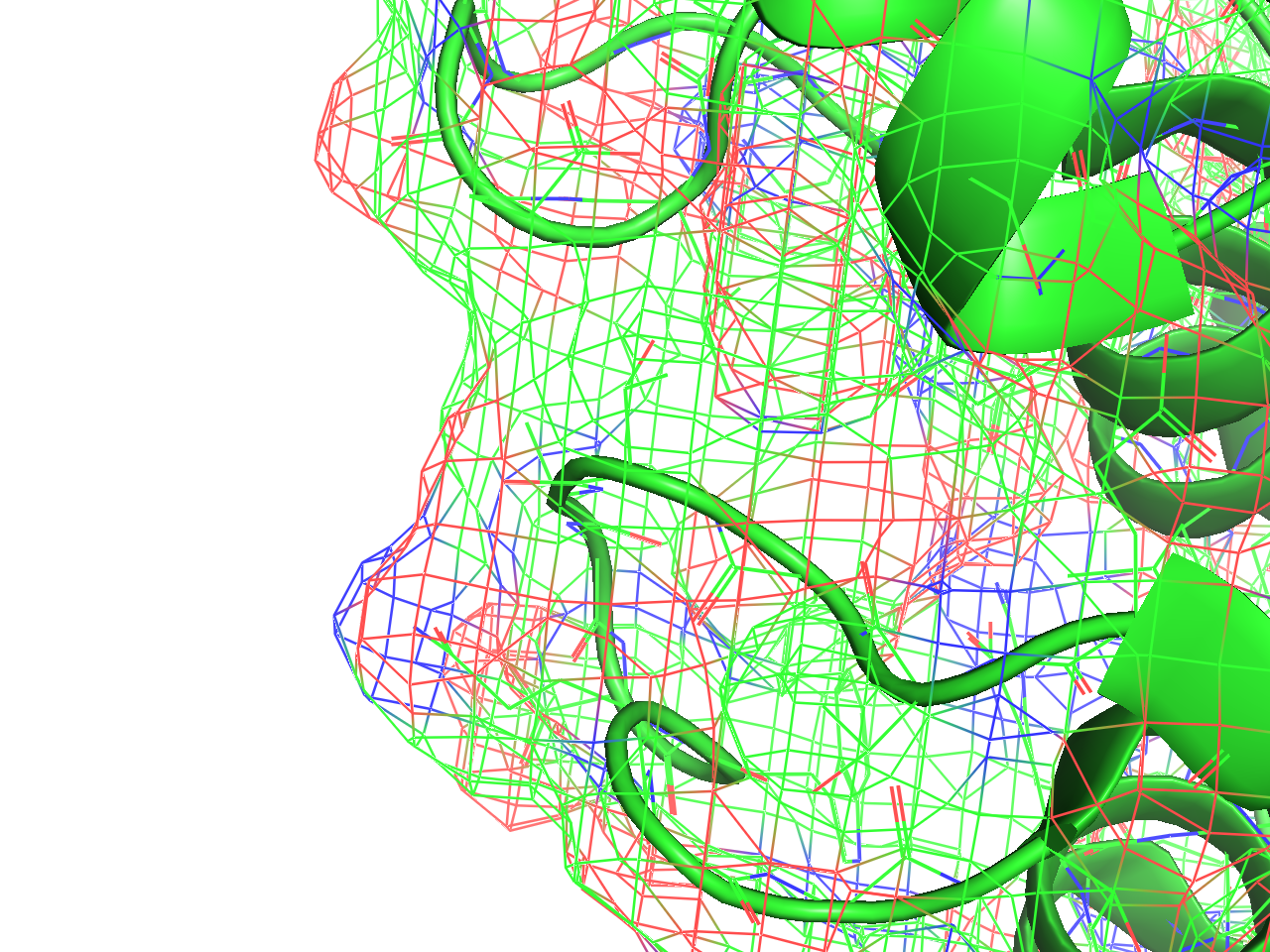
- Surface表示と似ていますが、表面の描画を網掛けで表現します。
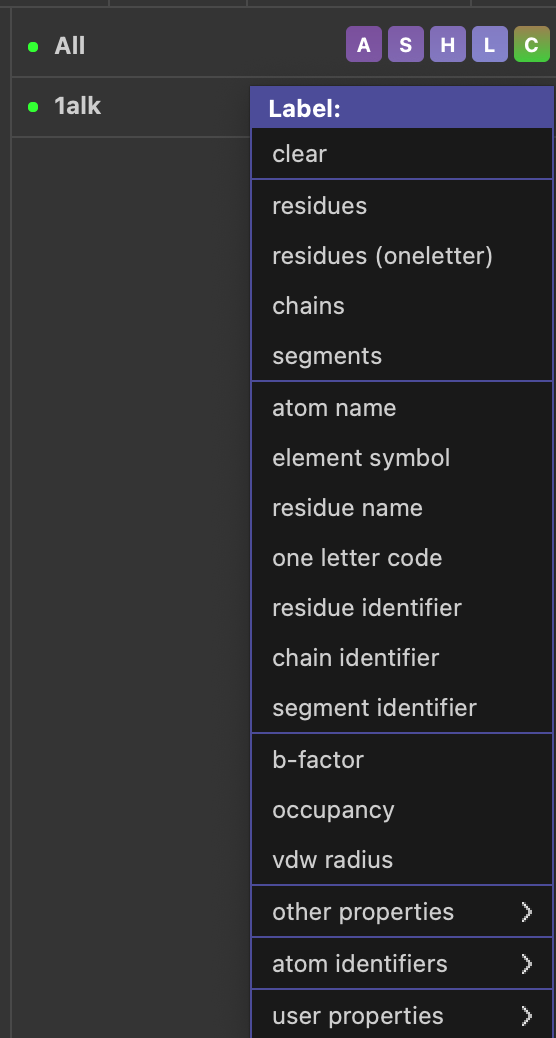
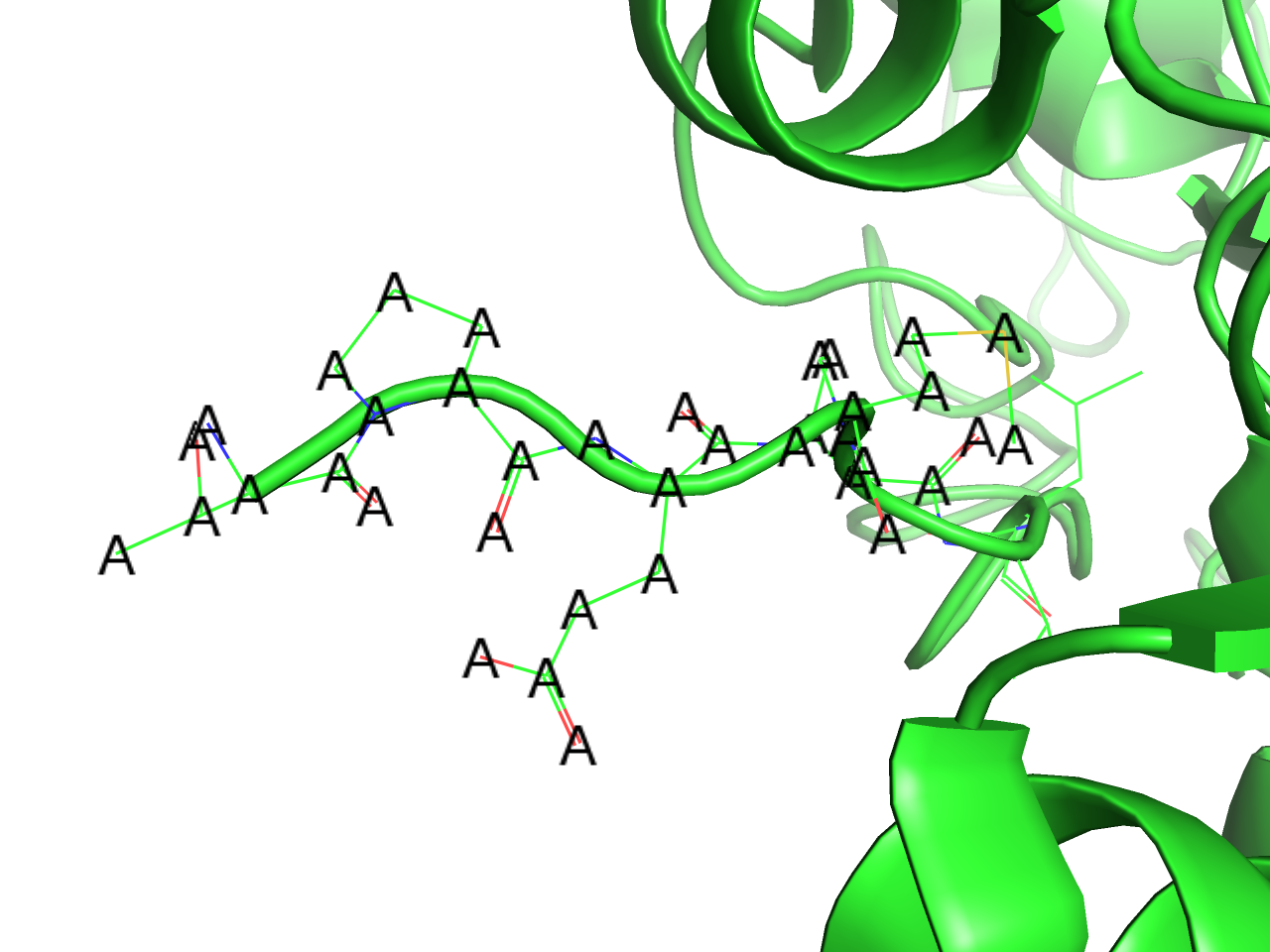
ラベルの設定(Label)
ラベルを利用することで、分子の周辺に文字を表示させることができます。活性部位周辺に存在する残基の名前と番号を表示させるだけでなく、任意の文字列を表示させることができます。
分子構造の表示形式のON/OFF (Show and Hide)のときと同様に、オブジェクトパネルから各オブジェクトに対して個別にラベルの設定を行うことができます。特に、選択範囲(sele)に対して表示させることが多いでしょう。
ラベルの設定変更はGUIの上部メニューから、Setting -> Labelのところにあり、ラベルの表示サイズやフォント指定、色の指定などが可能です。下記のサンプルではフォントサイズを28にしてあります。
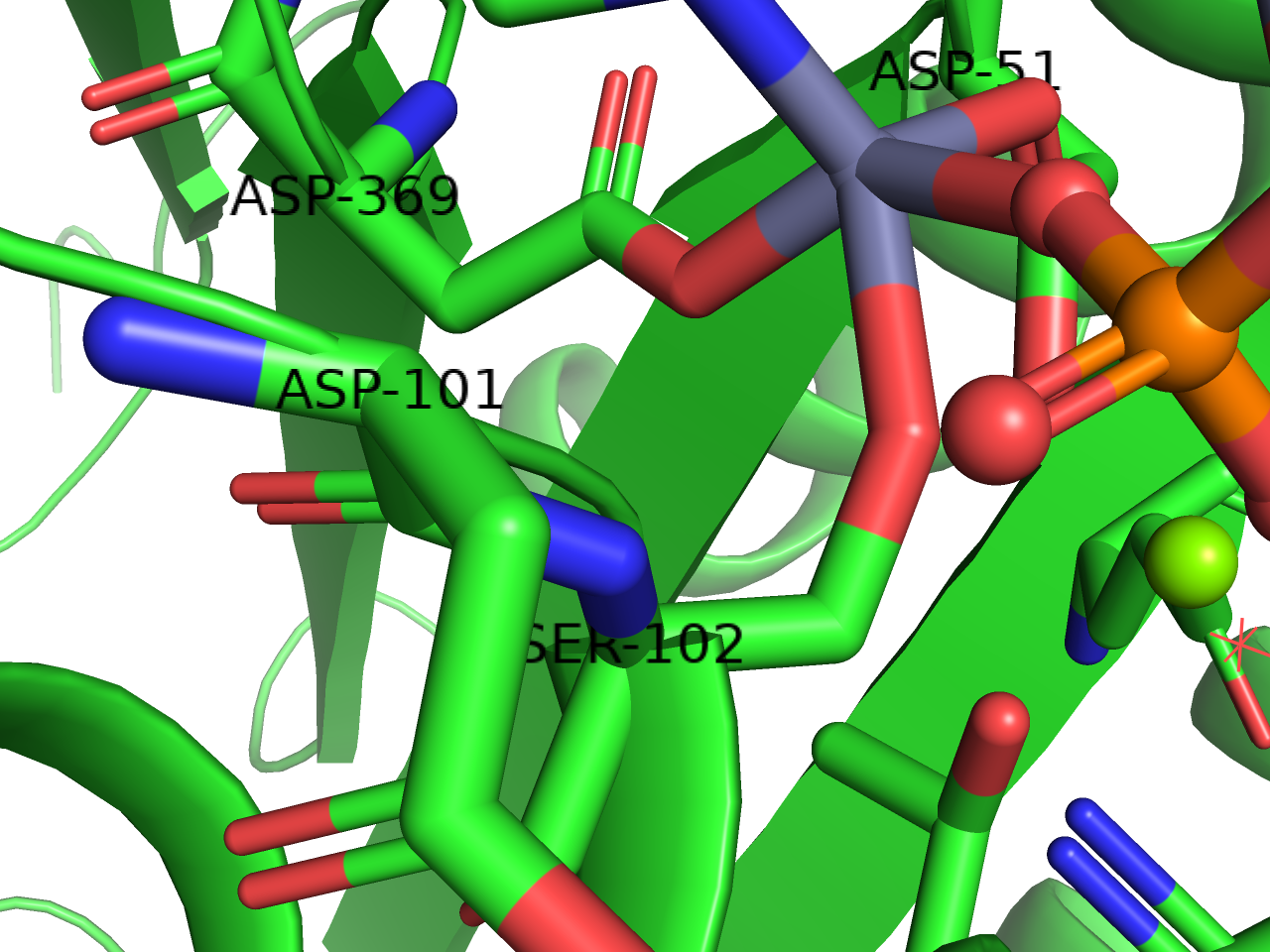
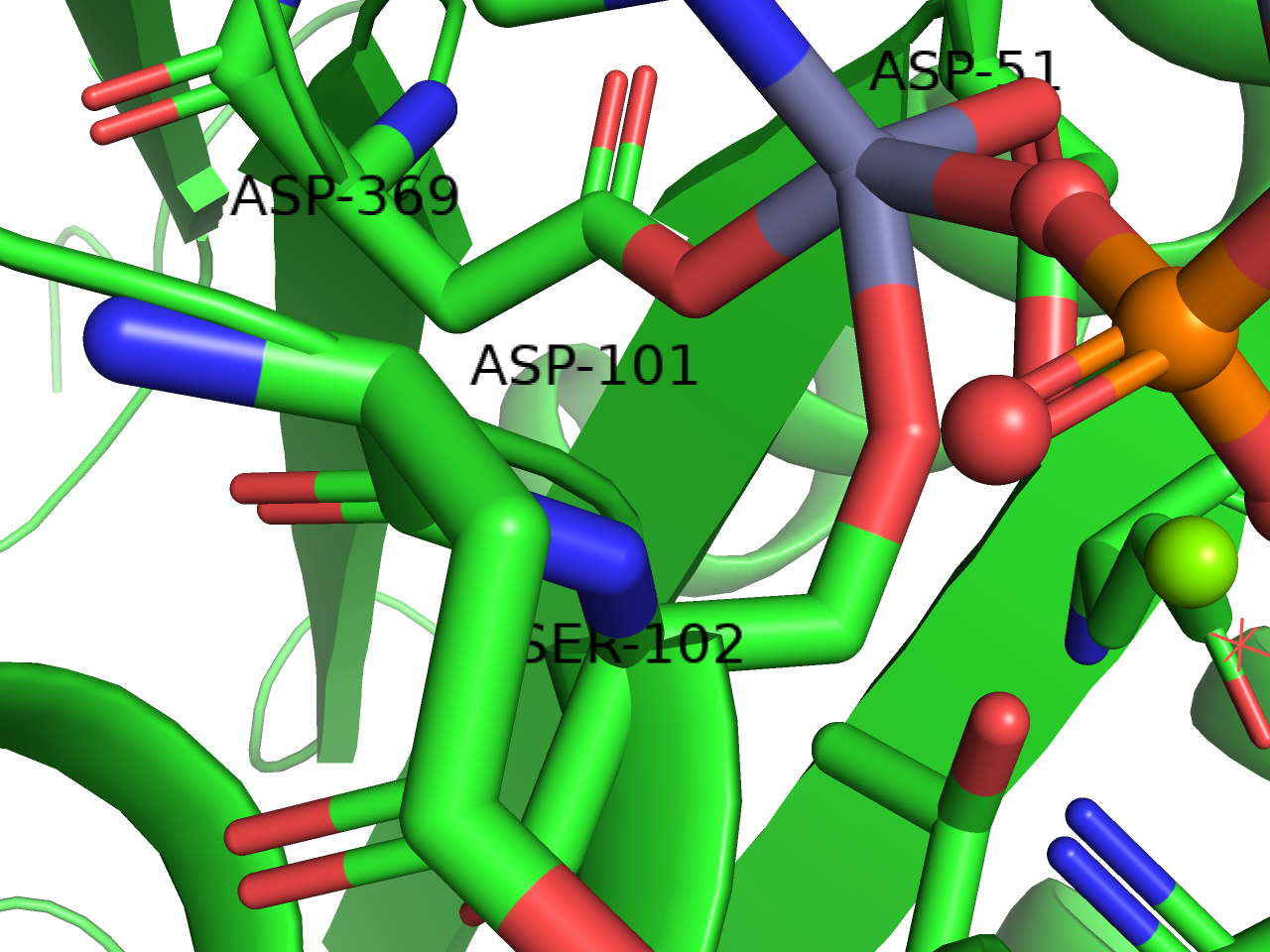
ラベルの位置変更は3-Button Editing Modeにしてから、ラベルの上でCtrl(macOSの場合はCommand)を押しながら左クリックでドラッグさせることで任意の位置に移動させることができます。下の例ではASP-101を右に移動させています。
clear
すでに表示されているそのオブジェクトについてのラベル表示を消去します。消去するのではなく表示を一時的に場合は、Hideメニューからlabelを選択しましょう。
residues
選択されたオブジェクトについて、アミノ酸と残基番号の情報を(アミノ酸3文字表記)-(残基番号)の形で表示します(例:MET-1)。Cα原子の上に表示される設定になっています。
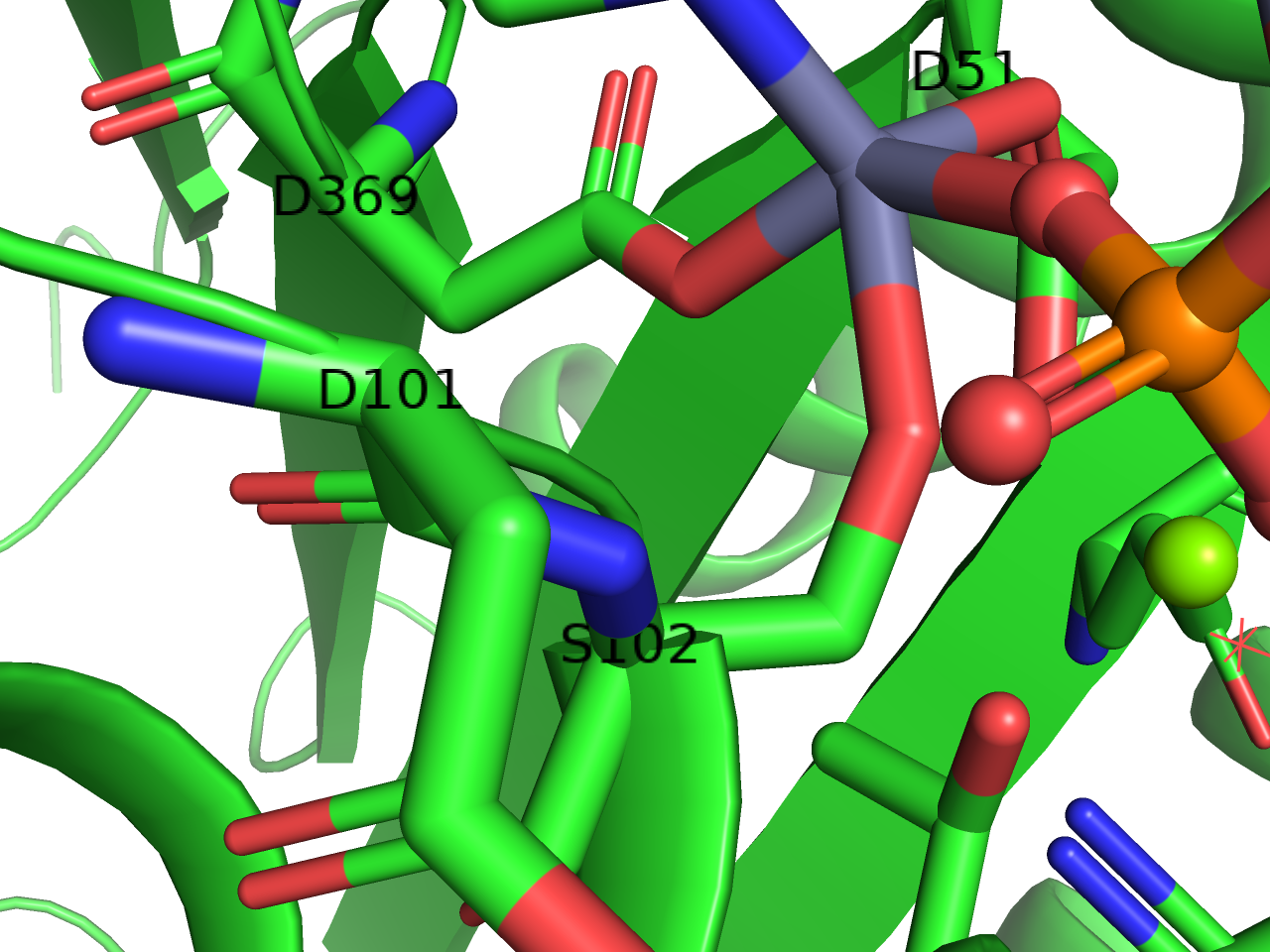
residues (one letter)
選択されたオブジェクトについて、アミノ酸と残基番号の情報を(アミノ酸1文字表記)(残基番号)の形で表示します(例:M1)。Cα原子の上に表示される設定になっています。
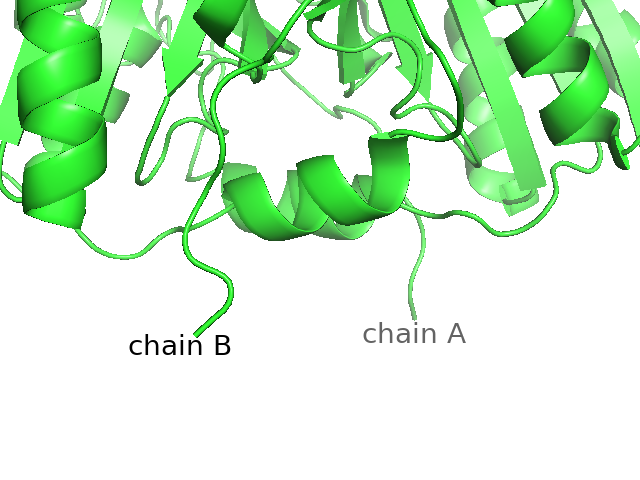
chains
選択されたオブジェクトのN末端とC末端の番号を持つ残基(リガンドが存在する場合、それも含む)のそばにchain <chain ID>の形で表示します(例: chain A)。
segments
すでに使用が廃止されたsegment identifierの情報を、chainsと同様に表示します。詳しくはAppendix: 用語#セグメントを参照してください。今となってはあまり使うことはないと思います。
atom name
原子名の名前でラベルを作成します。.pdb形式ファイルの場合、ATOM, HETATMレコードの13-16列目に含まれる文字列がatom nameとして定義されており、この文字列を返します。
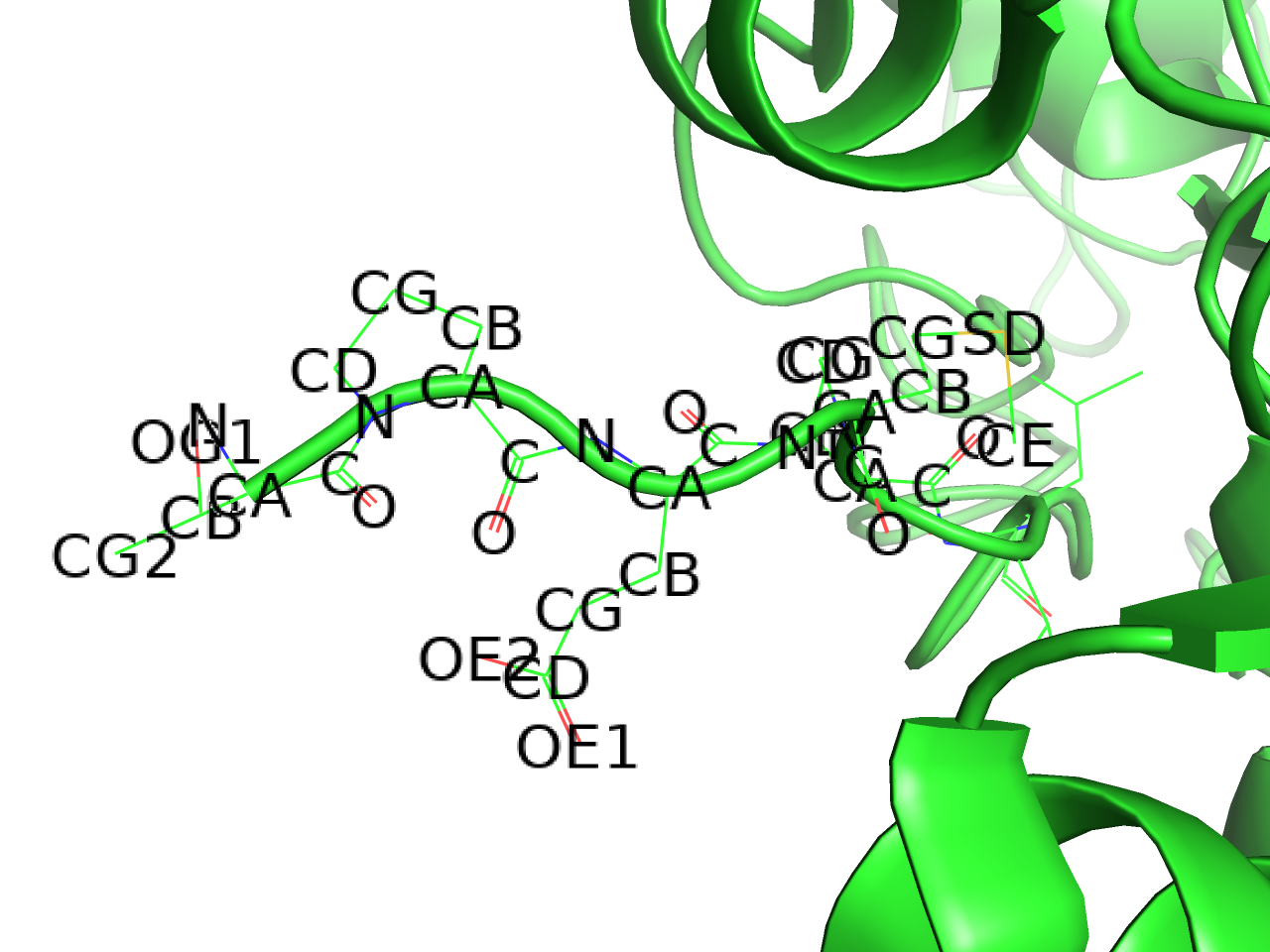
タンパク質アミノ酸やDNA, RNAの場合は命名規則があるため、慣れてしまえばatom nameの意味を理解しやすいと思います。13-14列目には右詰めで原子名を表します。例えば鉄原子は13-14列目を使ってFEの文字が入っていますが、炭素原子は14列目にCの文字を入れ、13列目は空白となっています。しかし、水素原子の場合は13-16列目の4文字分を最大限使って記述する場合があり、この場合は13列目から記入することになっています。
以下ではよく登場するatom nameの一例です。
| atom name | 意味 |
|---|---|
| CA | 主鎖のα位の炭素 |
| N | 主鎖のN末端側窒素原子 |
| C | 主鎖のC末端側炭素原子 |
| O | 主鎖のC末端側酸素原子 |
| CB | 側鎖のβ位の炭素 |
| CG | 側鎖のγ位の炭素 |
| CD | 側鎖のδ位の炭素 |
| CE | 側鎖のε位の炭素 |
| CZ | 側鎖のζ位の炭素(チロシンなど) |
| OH | 側鎖のη位の炭素(チロシンなど) |
主鎖に近い原子の個数順にβ→γ→δ→ε→ζ→ηと付ける命名規則があります。複数原子が存在するようなアミノ酸(ヒスチジン)などはND1, CD1, CE2, NE2というふうに番号をつけて区別します。
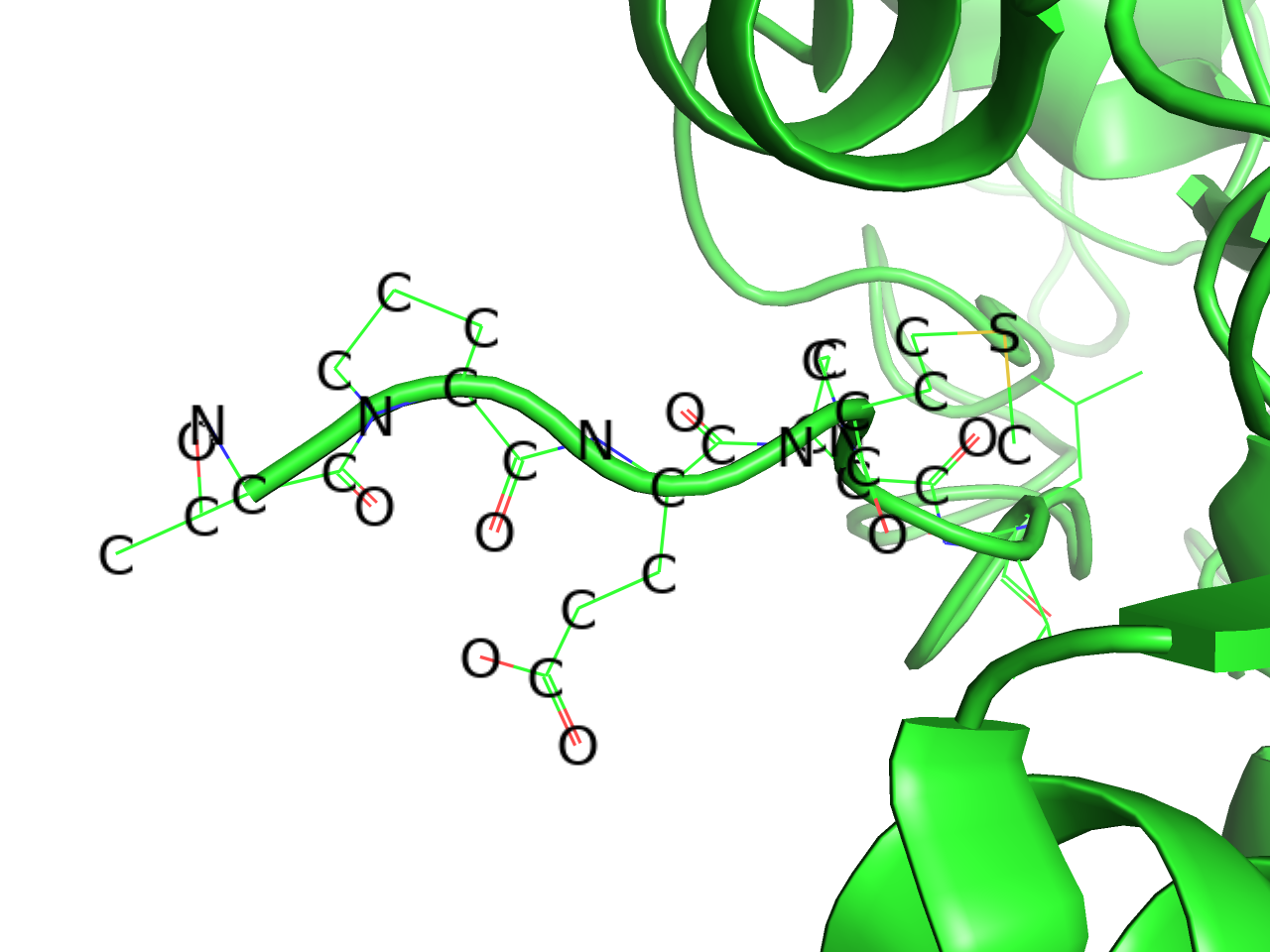
element symbol
原子名を表記します。
residue name
3文字表記の残基名を表記します。.pdbファイル形式におけるATOM, HETATMレコードの18-20列目の文字列を返します。存在しない場合は空白になります(以下同様)。

one letter code
1文字表記の残基名を表記します。
residue identifier
残基のresidue IDを表記します。.pdbファイル形式におけるATOM, HETATMレコードの23-26列目の文字列を返します。
chain identifier
残基のChain IDを表記します。.pdbファイル形式におけるATOM, HETATMレコードの22列目の文字列を返します。
segment identifier
残基のSegment IDを表記します。詳しくはAppendix: 用語#セグメントを参照してください。
b-factor
各原子の温度因子(b-factor; temperature facotr)を表記します。.pdbファイル形式における61-66列目の文字列を返します。

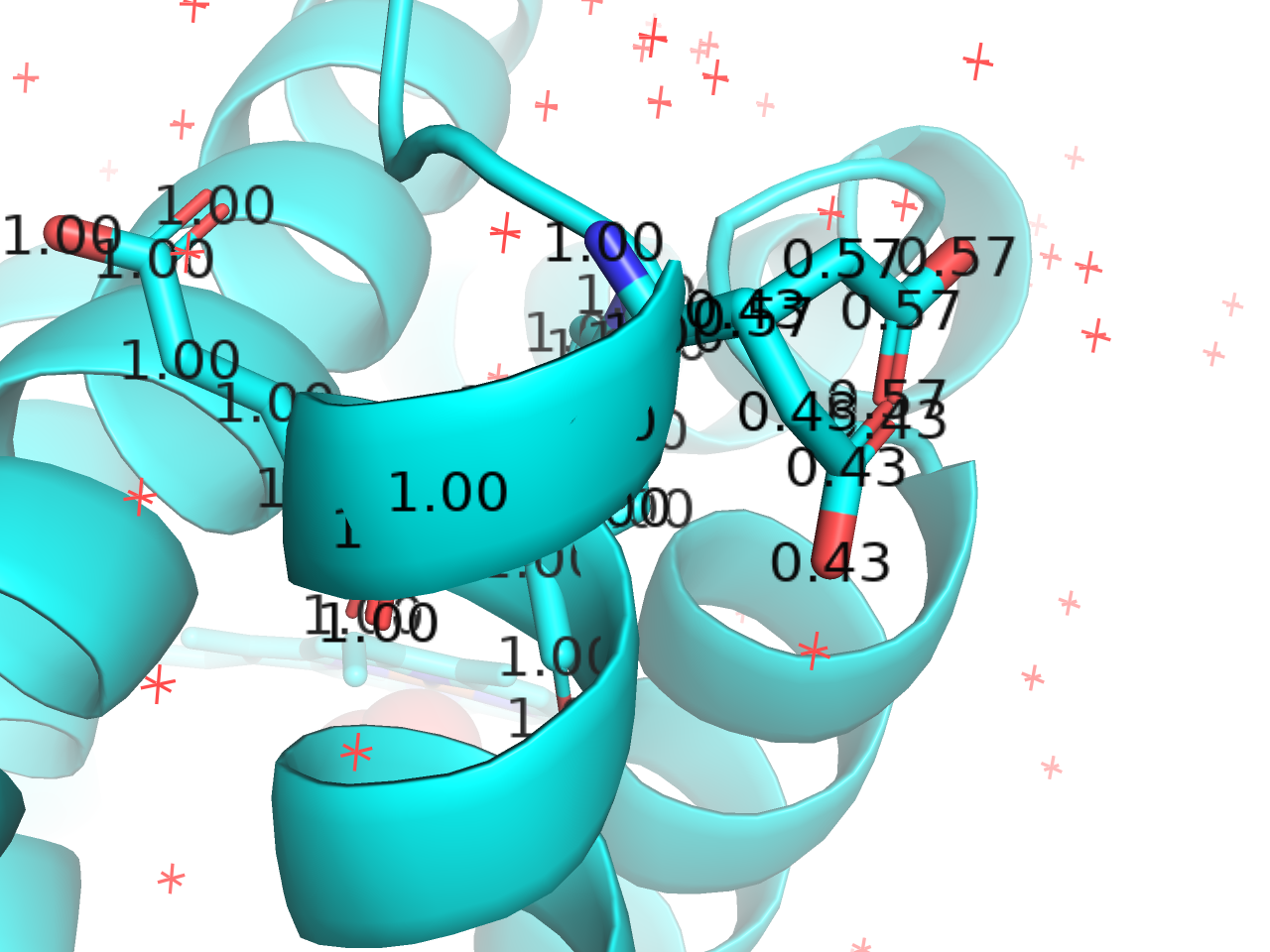
occupancy
各原子の重み因子を表記します。.pdbファイル形式における55-60列目の文字列を返します。結晶中では基本的に構造は繰り返しになるため、どの場所を見ても同じ座標がループしている事が多く、この値は1.00であることが多いのですが、時々その繰り返し構造単位の中に、1より大きなn通りの配座を持っていることがあります。このとき、ここには0~1の小数値が入ります。例としては PDB: 1A6MのGLN-8が0.57, 0.43の値を持つ2通りの配座を持ち、同様にTYR-151も0.50, 0.50で2通りの配座を持っています。この場合、PyMOLの表示上でも2通りの残基配座が見えます。画像はPDB: 1A6MのGLN-8の例を示しています。
vdw radius
各原子のファンデルワールス半径の値(Å)を表記します。基本的には原子の種類によって固定の値です。

Other properties
formal charge
各残基についてシステムで定義されている電荷を表記します。例としてLys, Arg残基の場合は+1を、Glu, Asp残基の場合には-1を表示します。
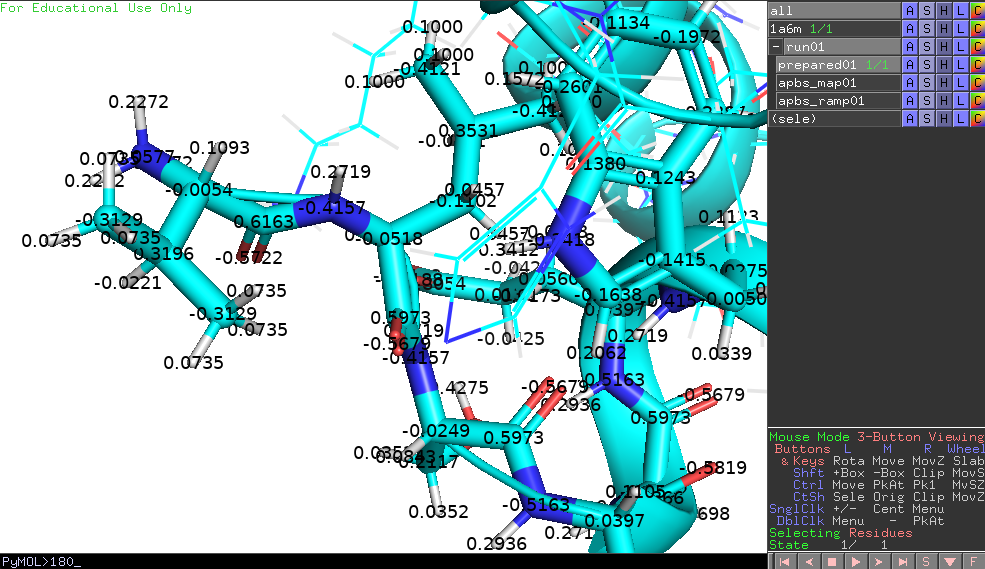
partial charge
各原子について計算された部分電荷(partial charge)を表記します。これは通常のpdb, mmCIFといった構造ファイルには含まれていないため、0.00と表示されてしまいますが、pqrやmol2ファイル形式をロードしたときに真価を発揮します。
タンパク質構造の場合は構造ファイルをいったんロードした後、APBSプラグインを使った表面電荷表示の手順に従って処理を進めます。このときにオブジェクトパネルにrun01->prepared01というオブジェクトが生成されるので、prepared01オブジェクトのラベル設定でpartial chargeを選択すると図のように部分電荷が表示されるようになります。
より正確に言えば、これはPDB2PQRというソフトウェアによって分子動力学シミュレーションソフトウェアAMBERが配布している力場AmberFFに基づいて指定されており、各アミノ酸の原子ごとに固有の値を指定してくれます。
実態としてはPQRファイルの記述様式にのっとり、x, y, z座標の次にくるchargeの値を表記しています。
mol2形式の場合でも対応しています。
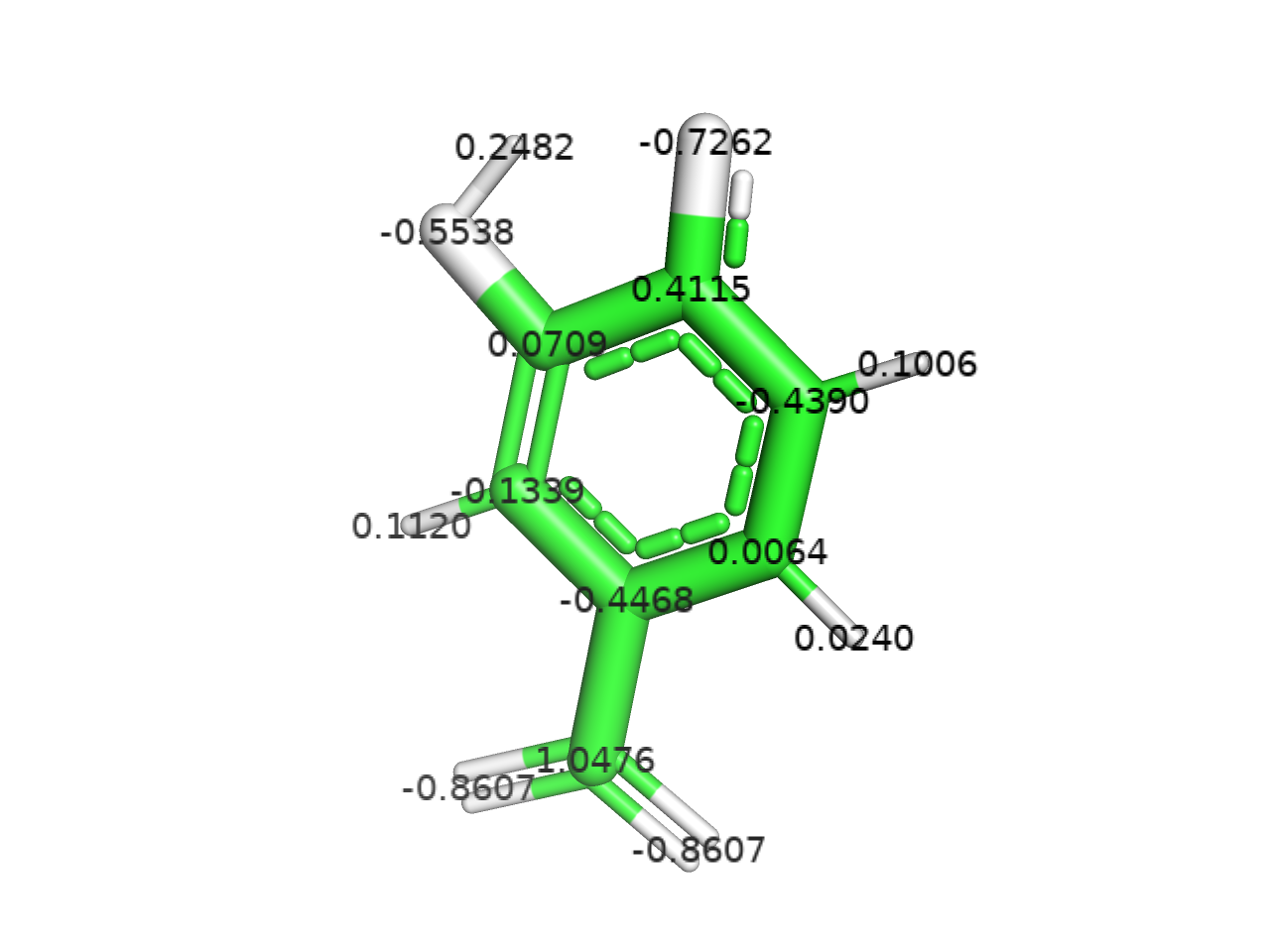
GaussianやGAMESSなどの量子化学計算ソフトウェアで計算した部分電荷を図にして見てみたいときに特に有効です。
elec. radius
partial chargeのときと同様に、PQRファイルのオブジェクトの場合に効果を発揮します。PQRファイル内のx, y, z座標, chargeの次にくるradiusの値を表記しています。vdw radiusとは異なります。
text type, numeric type
PyMOL 2.0以降は廃止されているらしく、使われていないようです(詳細不明)
stereochemistry
PyMOL 2.2から実装されました。schrodingerのソフトまたはrdkitモジュールのインストールが求められるようです。不斉炭素原子の部分にラベルを置いてくれる機能?
atom identifiers
rank
rankを表記します。rankとは構造ファイルの上から順にATOM, HETATMレコードを取得したときの通し番号のことであり、これのみ0始まりとなっています。
“rank” selects atoms based on their position in the source file, starting with zero as the first number.
ID
IDを表記します。IDとは構造ファイルから忠実に取得したATOM IDのことであり、pdbファイルフォーマットで言えば7-11列目に存在する番号のことを指します。この値は整数値でなければなりません。
“id” selects atoms based on the atom identifier number from the source file, which must be a integer.
index
Indexを表記します。indexはrankと似ていますが、順に表示するのとは異なり、PyMOL内部で番号を順に振り直したのがindexです。例えばある1つのアミノ酸残基があったとき、その残基の中で登場する原子の順番をどのように入れ替えても、index表記ではN→CA→C→O→CB→……の順に若い数字を付けていきます。
“index” selects atoms based on their internal PyMOL object atom indices starting with one as the first number.
色の設定(Color)
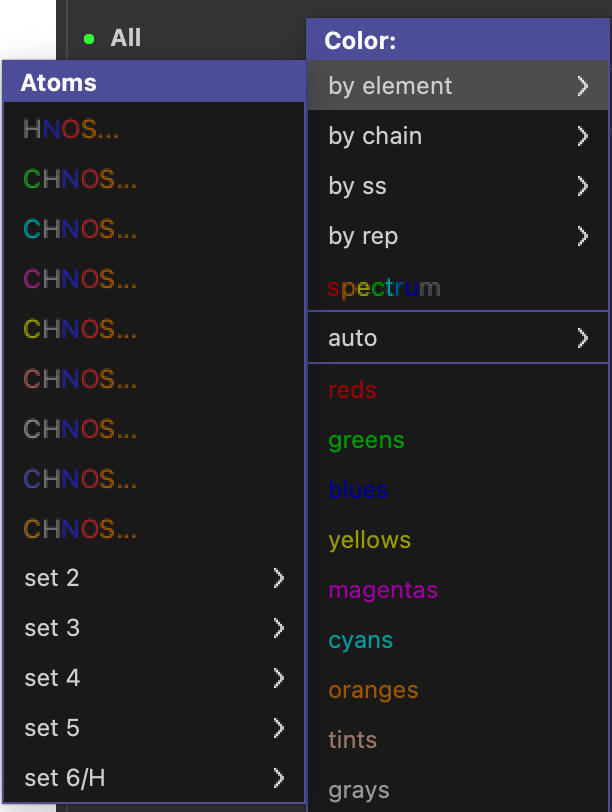
色の設定の仕方は自由です、センスの見せ所です……とは言うものの、実はタンパク質の構造を毎日見るであろう構造生物学専門の研究者にとっては、ある暗黙の了解があります。それらを踏まえた上で、PyMOLのカラーリング機能を紹介していきましょう。
色設定の暗黙の了解
以下のルールだけは遵守しましょう。
- 常に酸素原子は赤色, 窒素原子は青色, 硫黄原子はオリーブ(olive)色で表示する。
- 存在する場合、水素原子は白色で表示。
- よって炭素原子は、上記以外の色を使う。
メチオニン残基の表示の例(炭素原子はgreen)
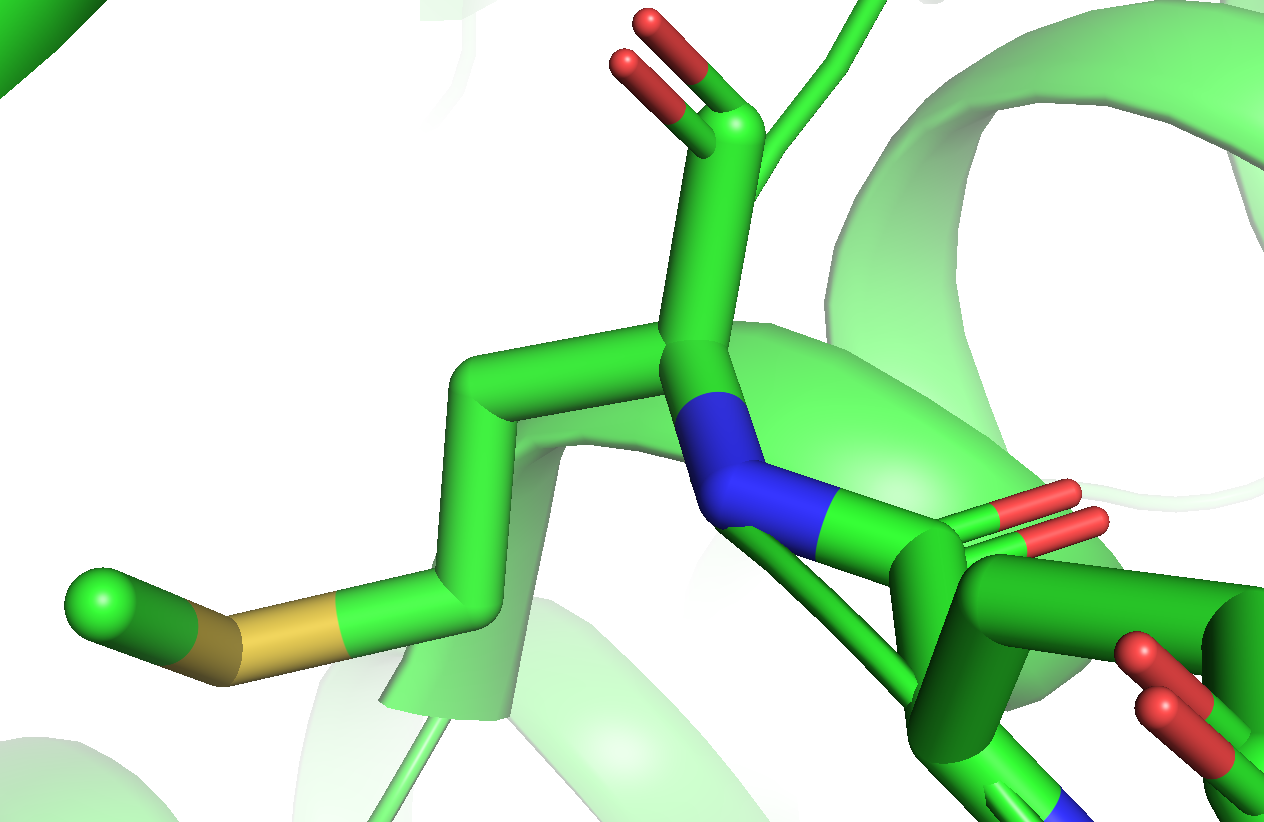
PyMOLには、酸素・窒素・窒素・水素原子を上記の色ルールで描画してくれるメニュー(“by element”)が存在しますので、これを使うことを強く推奨します。
正確には、PyMOLでは酸素原子はカラーコードで#FF7C7C (255, 76, 76) [1.0, 0.3, 0.3], 窒素原子は#3333FF (51,51,255) [0.2, 0.2, 1.0], 硫黄原子は#E7C640 (231, 198, 64) [0.9, 0.775, 0.25]がデフォルトで割り当てられているため、用意されているカラーパレットを使う限りにはこれらの色と完全にかぶることはないのですが、それでもこれらに近い色を使うと紛らわしいので、使用は控えましょう。また、以下で見るように、用意されているカラーパレットの中にはすべての原子を同一で塗るものもあるので、どうしてもそれらの色を使いたい場合は、そのカラーパレットを選択した後に“by element“メニューの一番上のカラーリング(通称“color by hetetoatom”)を選択してあげましょう。
by element
メニューを開いていただけると一目瞭然、C(炭素)の色だけが変わっており、H, N, O, S, 他にも様々な原子について、「暗黙の了解」の色で固定されています。反対に炭素原子の色を自由に選べることで、多量体タンパク質や大きな複合体に対しても自由に色を塗り分けられることができます。基本的には、このカラーパレットから選びましょう。
見落としがちですが、下の方にあるset 2〜set 6にも、様々な色が用意されています。set 6のみ水素原子の色が白色でないものが用意されていますが、あまり使うことはないでしょう。
一番上のメニューではCが表示されておらず、HNOSと書かれていますが、これは炭素以外の原子の色を既定色に変更するというとても便利なカラーパレットです(“color by heteroatom”)。
コマンド入力はこちら(オブジェクト1alkについて行う場合)
util.cnc 1alk # color by heteroatom
by chain
by chain (elem C | */CA, | 無印)
`1つの生体分子オブジェクト内に複数のchainが存在している場合、各chainごとに異なる色で塗り分けてくれます。
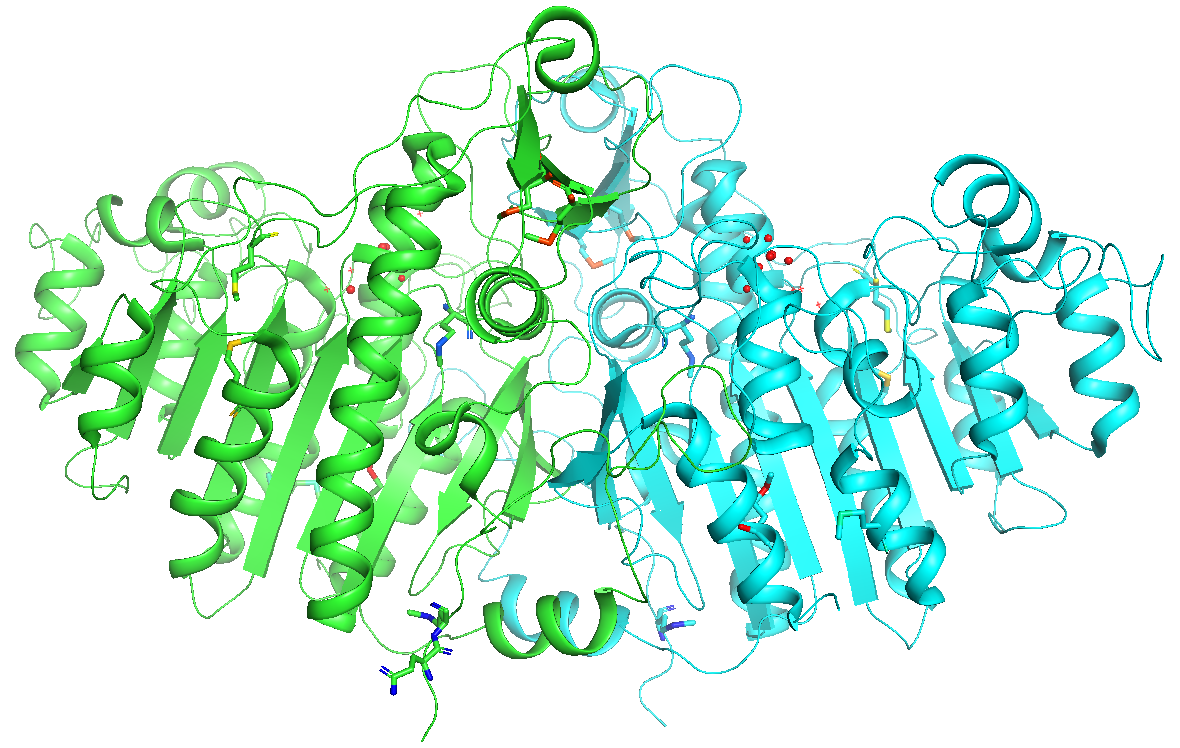
elem Cの場合、チェイン内の全炭素原子に対して色の変更を行い、他の種類の原子には色の変更を行いません。*/CA の場合、主鎖のCα原子のみ色の変更を行い、残りの原子には色の変更を行いません。無印は全原子に対して色の変更を行います。
コマンド入力はこちら(オブジェクト1alkについて行う場合)
util.color_chains("(1alk and elem C)") # elem C
util.color_chains("(1alk and name CA)") # */CA
util.color_chains("(1alk)") # 無印
chainbows
各chainについて、N末端(5’末端)からC末端(3’末端)にかけて青色から赤色のきれいなグラデーションができるように色を塗り分けてくれます。タンパク質の場合、この配色ならば、表示された色からアミノ酸配列の位置と構造上での位置の対応関係を把握しやすいため、この配色になっている論文を見たことがある方も多いでしょう。
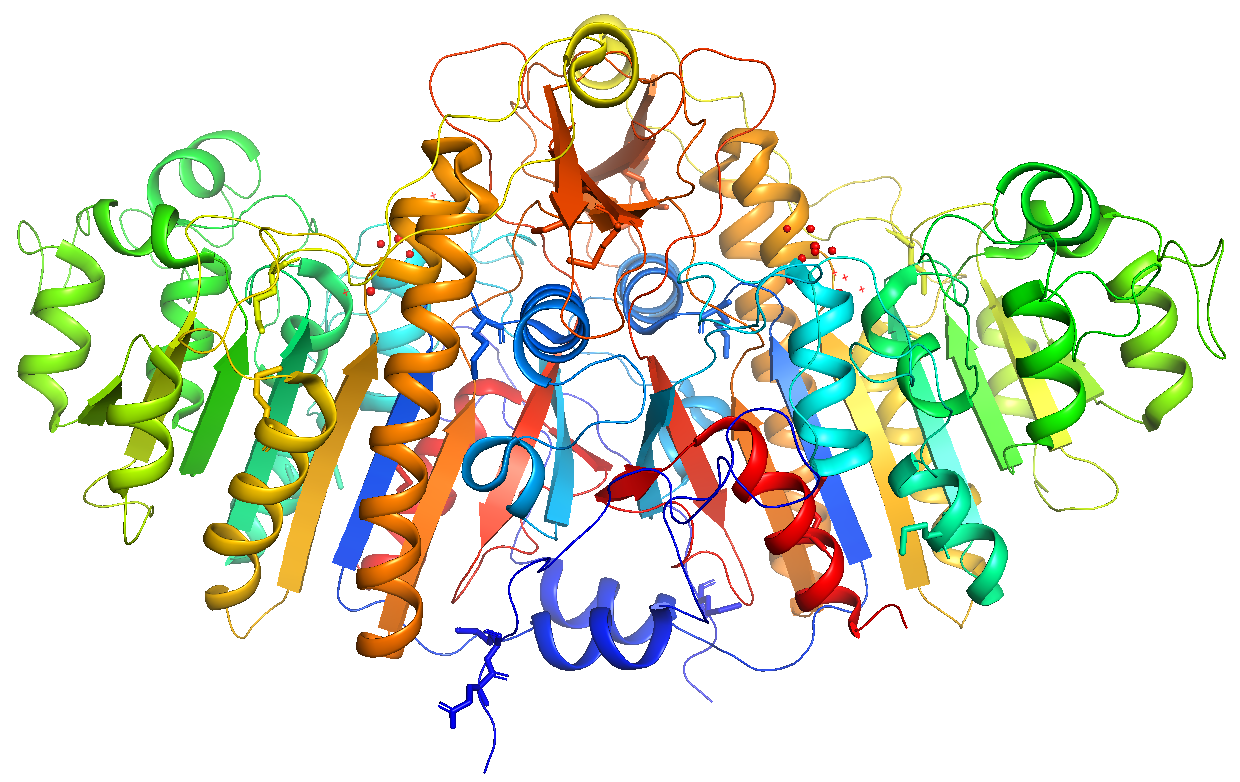
コマンド入力はこちら(オブジェクト1alkについて行う場合)
# どちらでもOK
util.chainbow 1alk
util.chainbow("(1alk)")
by segi (elem C | 無印)
segment identifierによる色分けを行います。
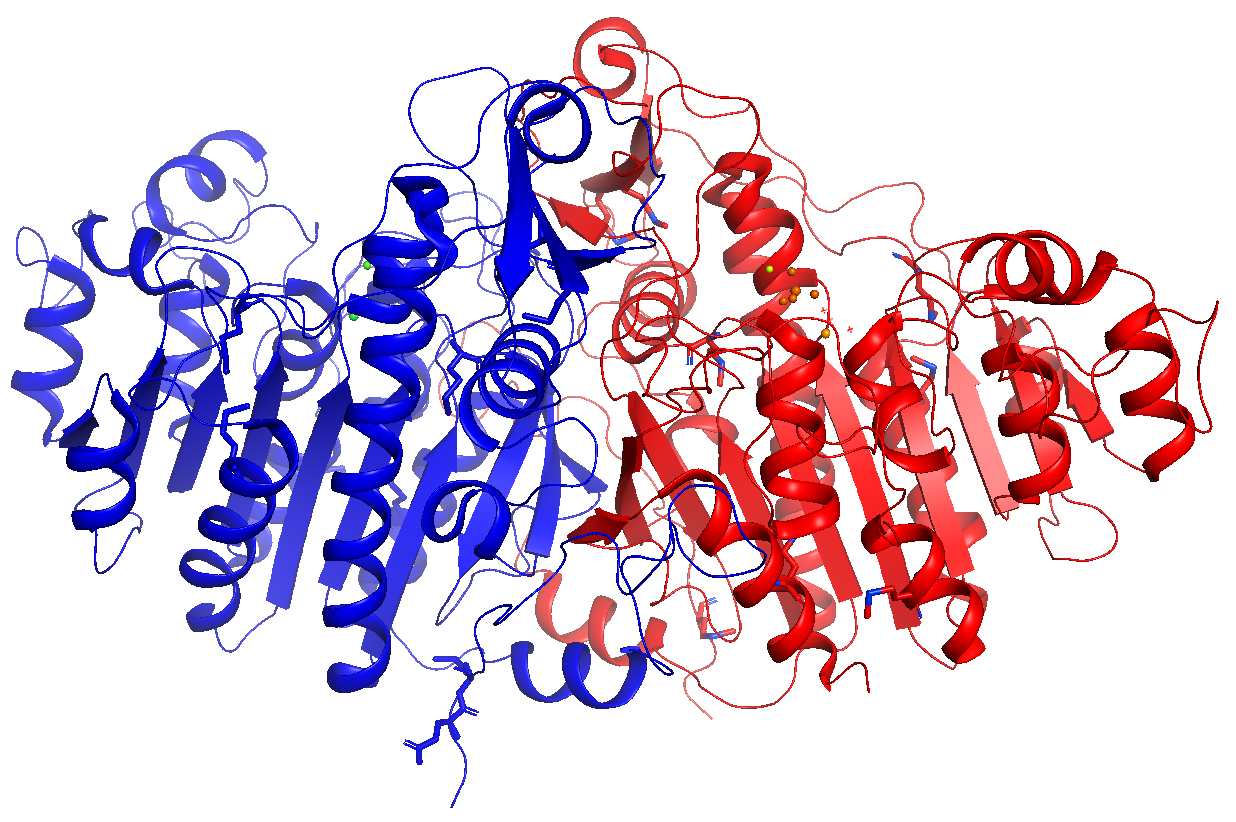
あまり詳しく調べていないのですが、segment identifierは現在仕様が廃止されつつあるのと、カラーリングが赤・青なので、あまり使うことはないでしょう。
by ss (By Secondary Structure)
タンパク質の二次構造(ヘリックス・シート・ループ)構造を基準に色分けしてくれます。自由に選びましょう。
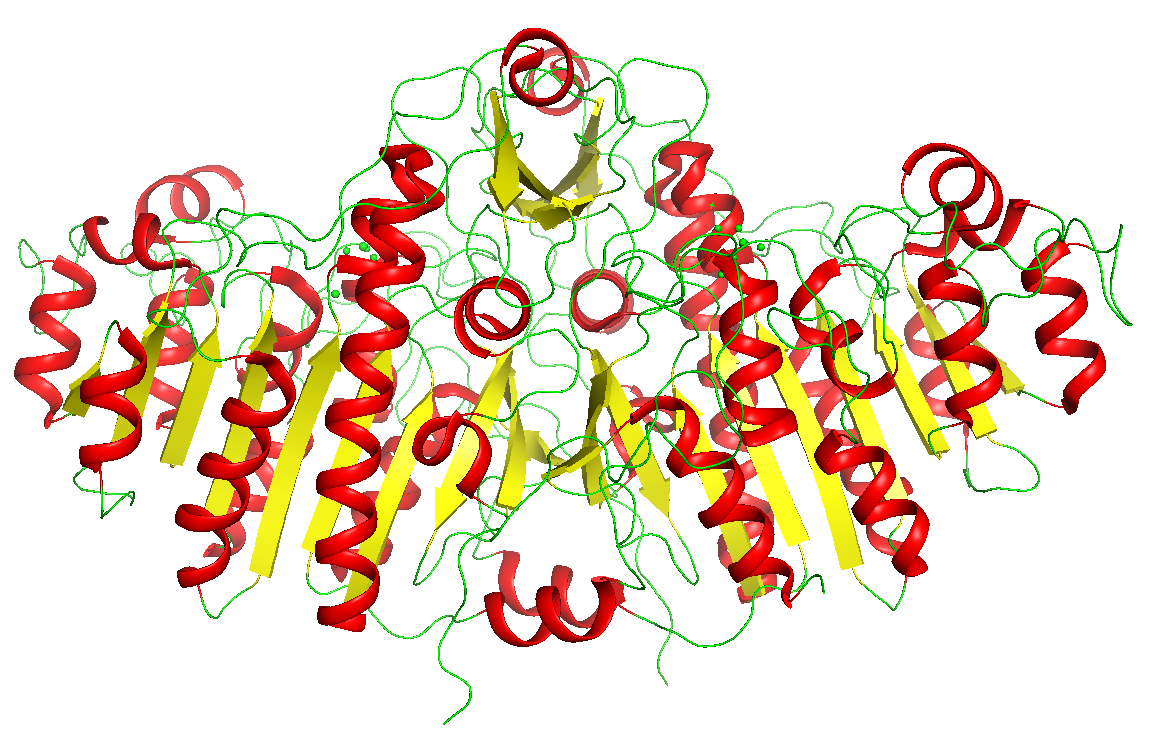
コマンドで行う場合で、ヘリックスを赤、シートを黄、それ以外を緑に塗りたいときは、以下のように打てばOKです。
color red, ss h
color yellow, ss s
color green, ss l+''
または、
util.cbss("1alk","red","yellow","green") # オブジェクト1alkについて行いたい場合
by rep (By Representation)
表示形式(representation)ごとに色分けを表示させることができます。これ以外の他のメニューからの色変更は、全表示形式が一括で色変更されてしまうので、うまく使い分けましょう。例えば、by chainで緑・青に色分けをした後に[show]→[surface]としてsurface表示をした状態ですと、
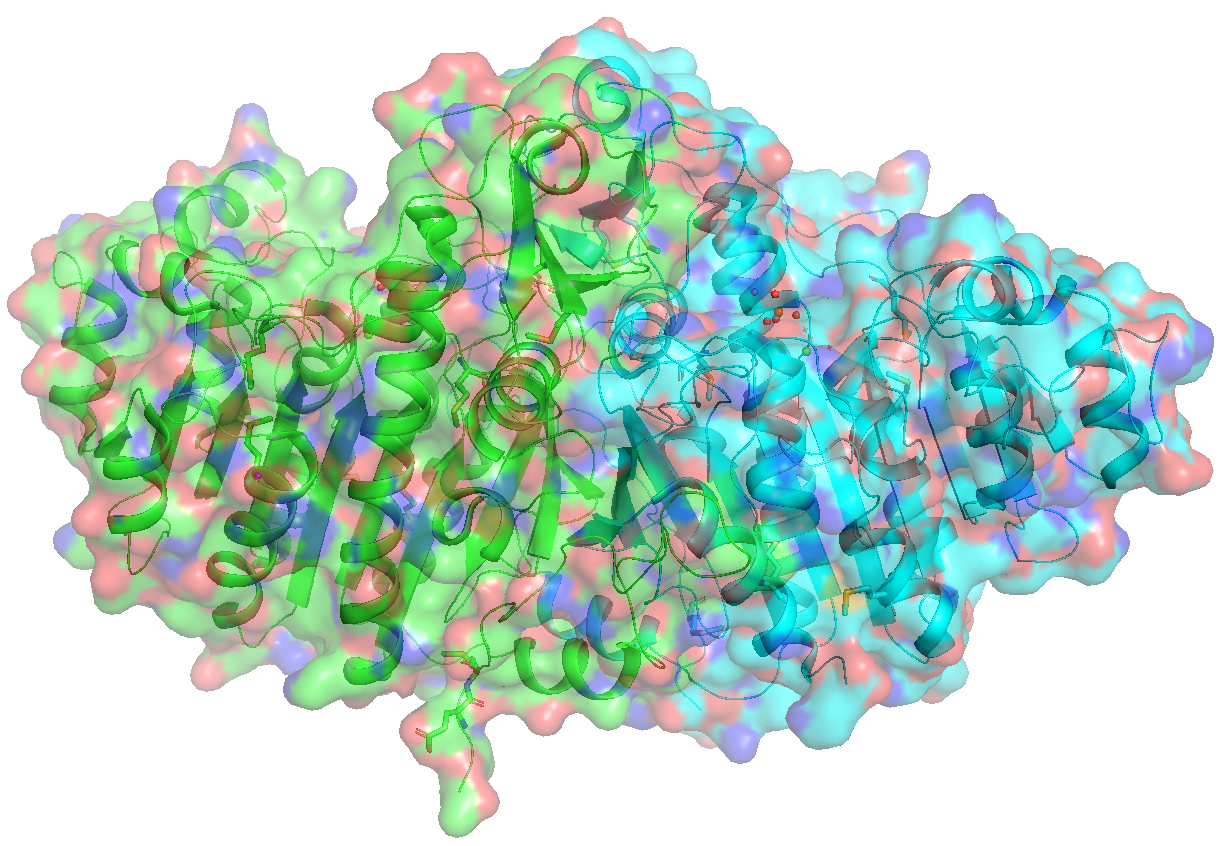
という表示になりますが(コマンドでset transparency, 0.5とし、Surface表示の透明度を変更しています。)、続いて[by rep]→[surface]→[grays]→[white]とすると
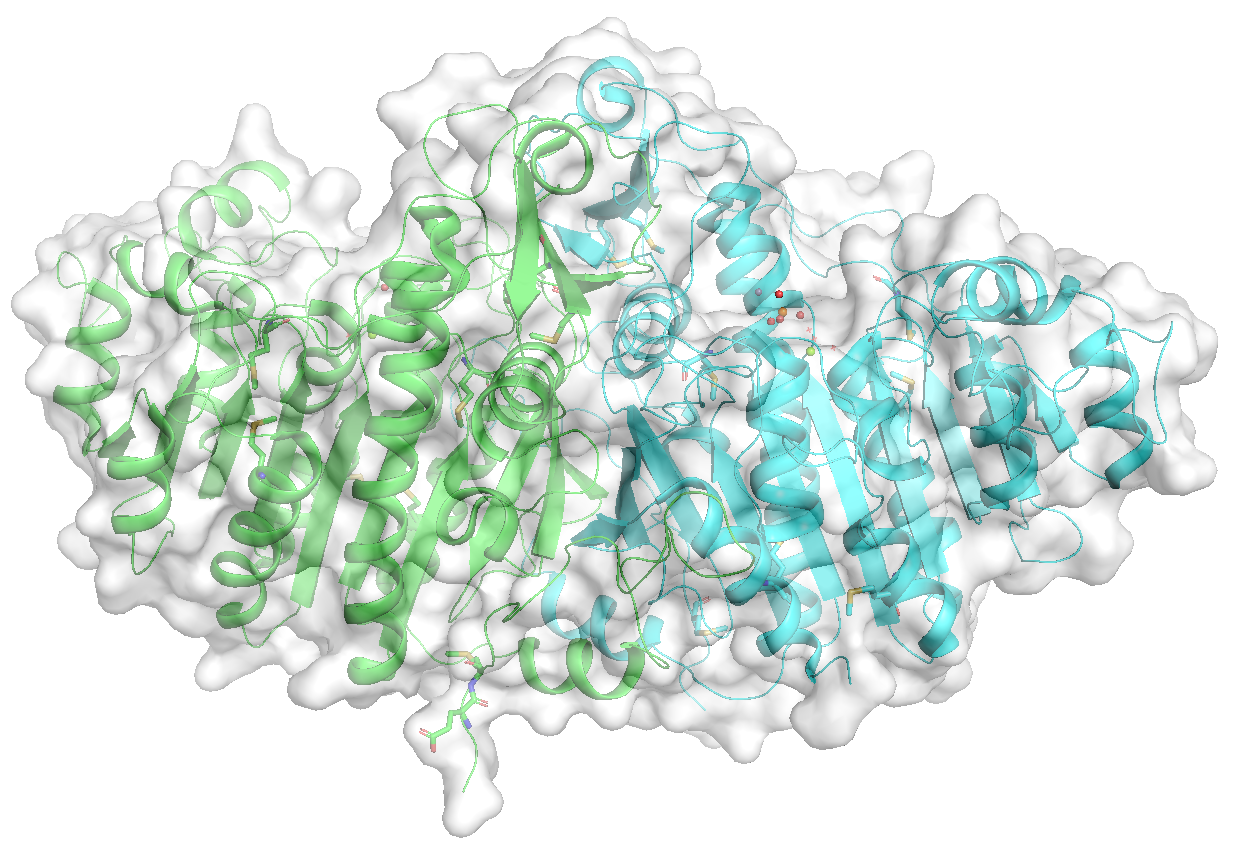
このようにSurface表示だけ白くすることができます。
コマンド入力はこちら(オブジェクト1alkについて行う場合)
# どちらでもOK
set surface_color, white, 1alk
cmd.set("surface_color", "white", '1alk', quiet=0)
quiet=1 とすると処理終了時にメッセージがログ欄に表示されなくなります(プログラミング用)。surface_color の部分をlineやcartoonなどに変更すればその表示形式に関わる色のみ変更できます。
spectrum
rainbow (elem C | */CA, | 無印)
オブジェクト全体を虹色表示してくれます。[by chain]の[chainbow]と違い、chainごとに虹色グラデーションをつけてくれるわけではないことに注意してください。elem C, */CA, 無印の意味は上と同様です。
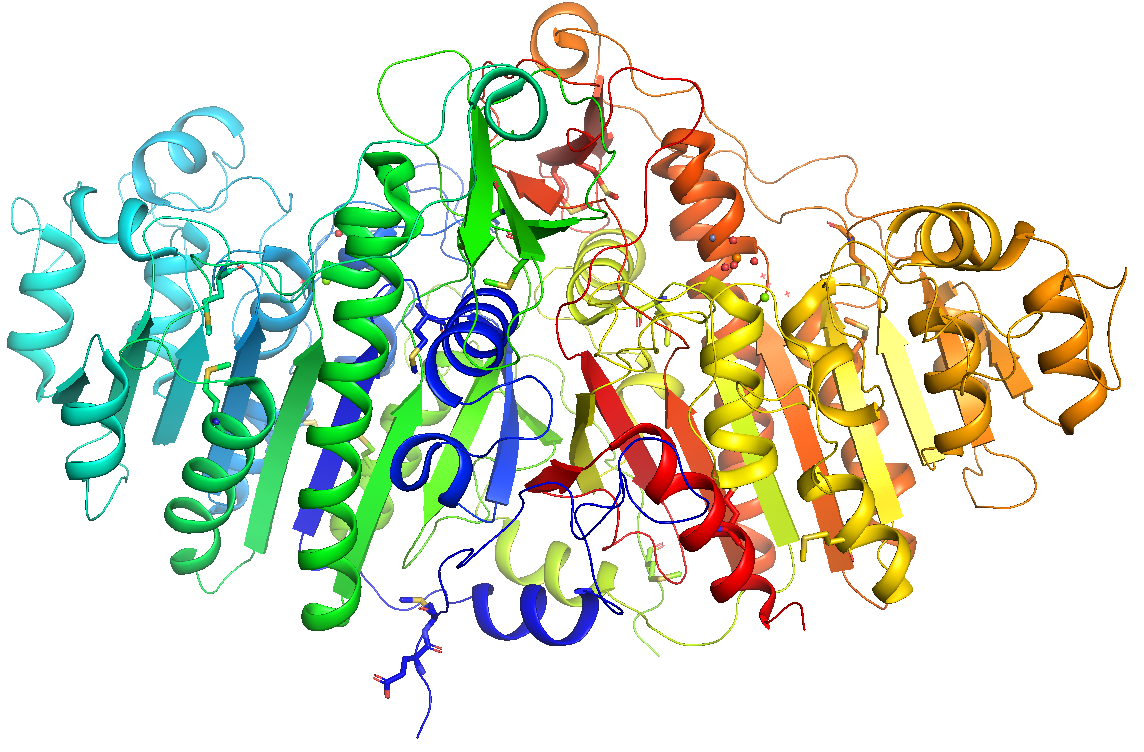
コマンド入力はこちら(オブジェクト1alkについて行う場合)
# elemCの場合。どちらでもOK。
spectrum count, selection=1alk and elem C
cmd.spectrum("count",selection="(1alk)&elem C")
# */CAの場合
spectrum count, selection=1alk&*/CA
cmd.spectrum("count",selection="(1alk)&*/CA")
# 無印
spectrum count, selection=1alk, byres=1
cmd.spectrum("count",selection="1alk",byres=1)
b-factors (無印 | */CA)
結晶構造解析法で構造決定された構造ファイルの場合、b-factor(温度因子) という値が各原子に対して割り当てられています。簡単に言えば、この値は その原子のゆらぎやすさ を表しており、概して分子内部に存在している原子ほど値が小さく、外側に存在する原子ほど大きくなっています。この色表示ではb-factorの値が小さいほど青色に、大きいほど赤色になるよう表示されます。
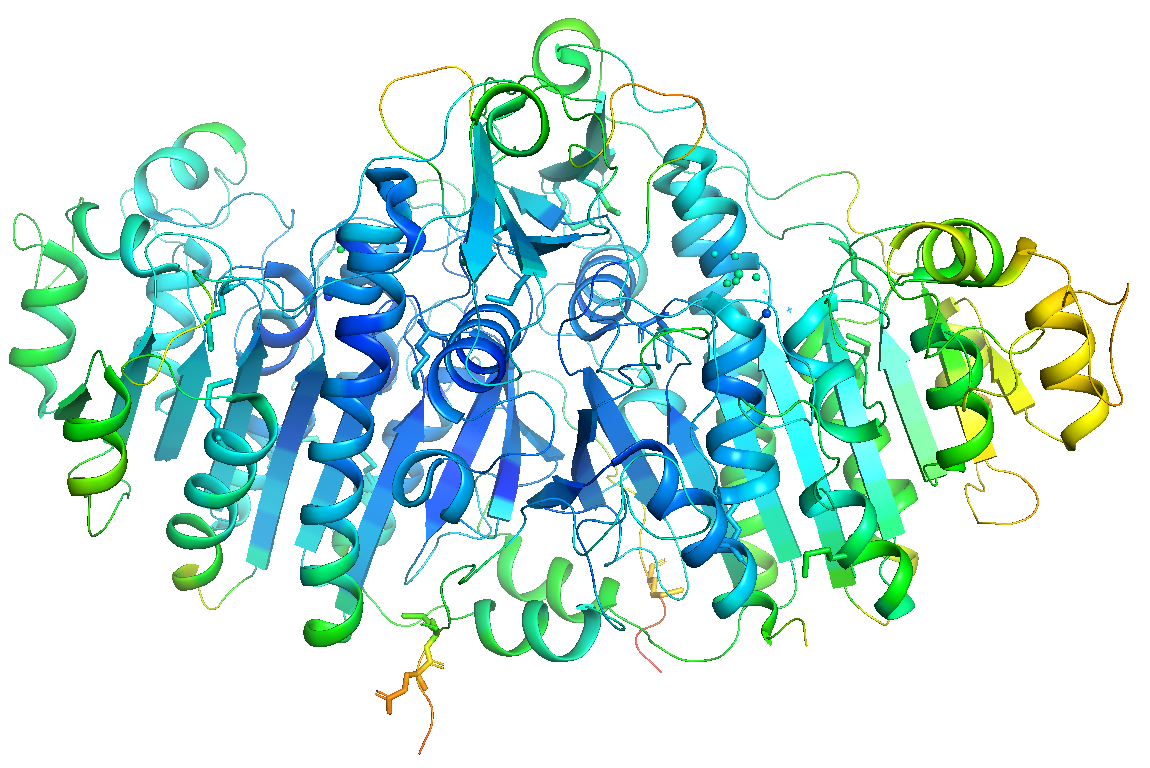
コマンド入力はこちら(オブジェクト1alkについて行う場合)
# 無印の場合。どちらでもOK。
spectrum b, selection=1alk, quiet=0
cmd.spectrum("b", selection=("1alk"), quiet=0)
# */CAの場合。どちらでもOK。
spectrum b, selection=1alk&*/CA, quiet=0
cmd.spectrum("b", selection="((1alk)&*/CA)", quiet=0)
表示形式のプリセットの中には、b-factorごとに色と太さを変えてくれるオプションもあります。
area (molecular | solvent)
残基あたりのsurface area(表面積)を計算し、それに応じた値でグラデーション色付けをしてくれます。
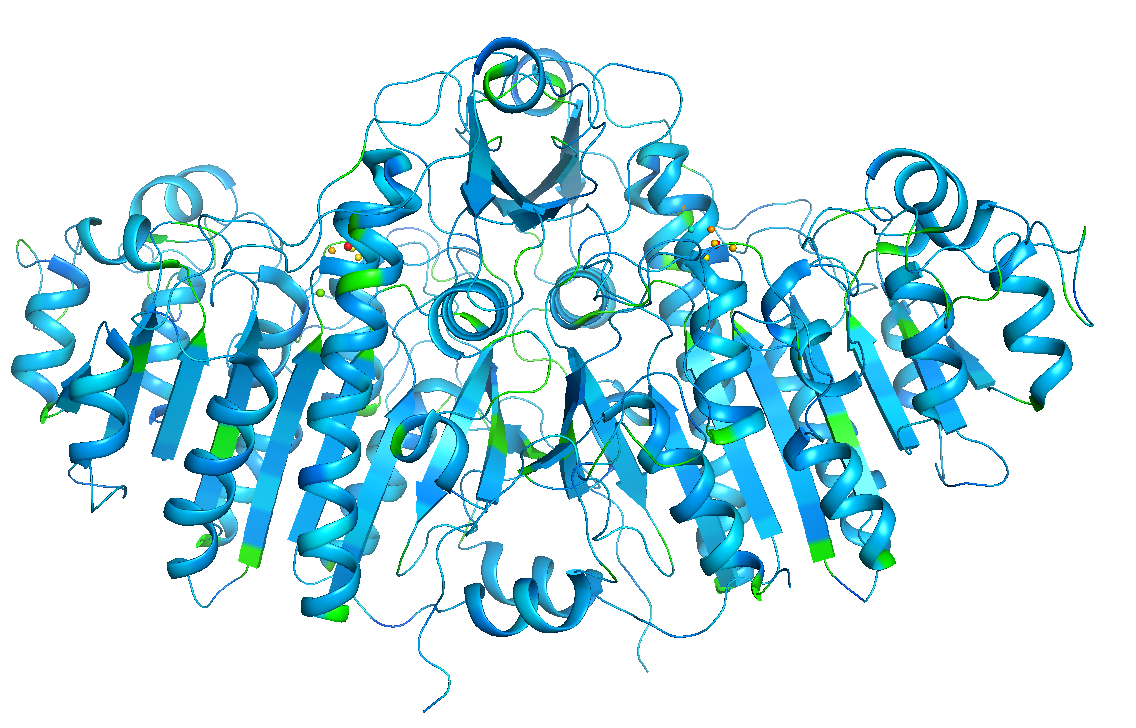
プログラムの仕様上はget_areaコマンドを走らせて、その値に応じた色付けを行っているようです。しかし、solventの方も含めて、かなり雑な計算なのでまったく厳密ではないようです(参考:https://pymolwiki.org/index.php/Get_Area)。Solvent accessbile surface areaを計算したい場合は、別の厳密なソフトウェアで計算した値を使うことを推奨します。
コマンド入力はこちら(オブジェクト1alkについて行う場合)
# molecularの場合(デフォルト)。
util.color_by_area("1alk","molecular")
# solventの場合
util.color_by_area("1alk","solvent")
auto (elem C | all | by obj (elem C) | by obj)
自動で色付けをしてくれます。複数の生体分子オブジェクトがPyMOL上で開かれている場合に有効です。上2つは、押すたびに色が入れ替わっていきます。緑→シアン→黄→紫……の順に入れ替わります。

下2つのby obj がついているものはロードされている生体分子オブジェクトの上から順に色を緑→シアン→黄→紫……の順で塗り分けてくれます。特に、all を選択してby obj を押せば一発で見慣れた色分けになってくれるので便利です。
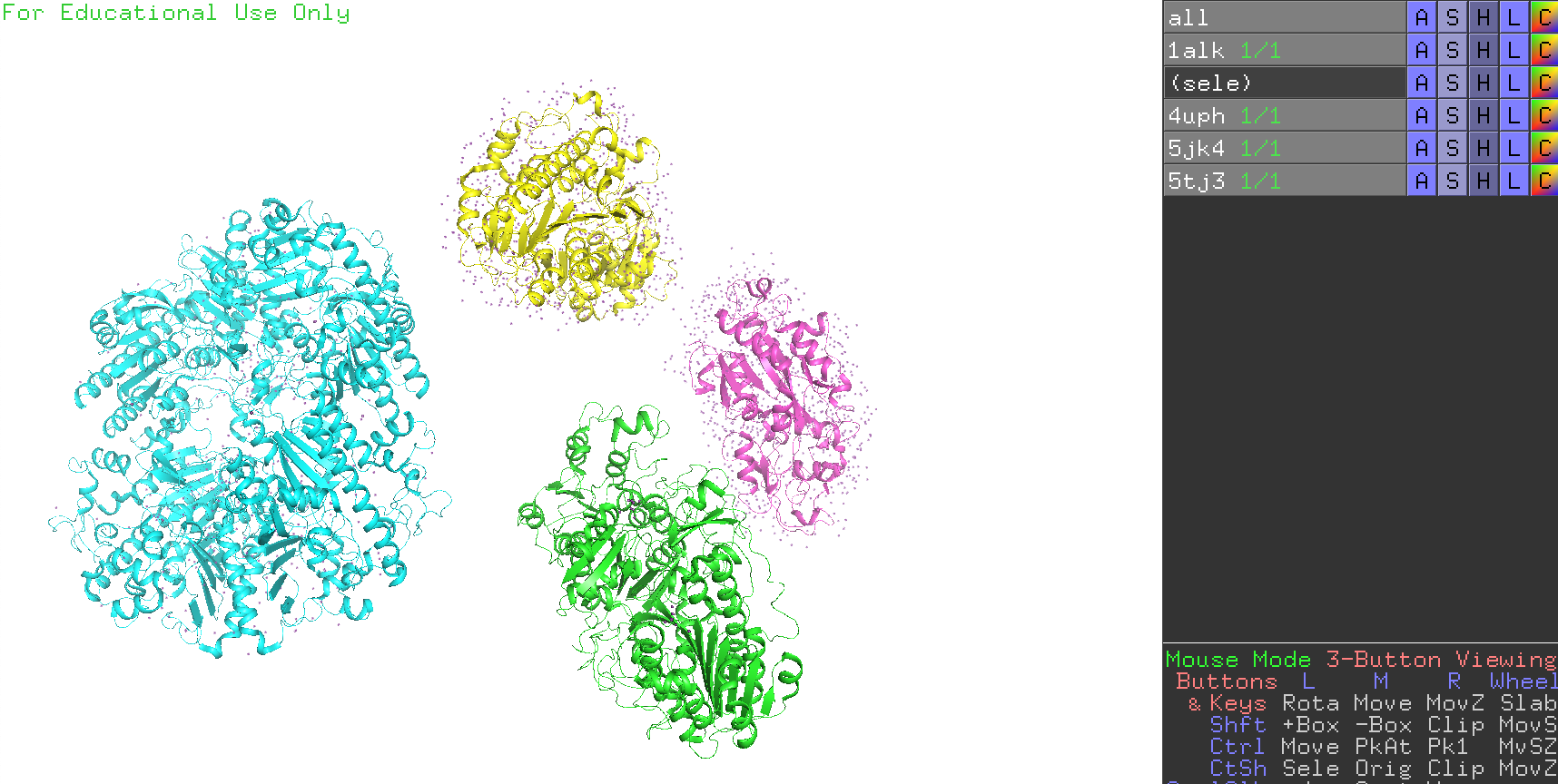
コマンド入力はこちら(全オブジェクトについて行う場合)
cmd.color("auto","(all) and elem C")
cmd.color("auto","all")
util.color_objs("(all and elem C)",_self=cmd)
util.color_objs("(all)",_self=cmd)
auto以下に存在する色分けメニューについて
autoメニューの下には様々なカラーパレットがあります。自由に選択して好きな色を選びましょう。全表示形式(line, stick, surface…)について同一の色を適用します。
コマンド入力はこちら(オブジェクト1alkについて赤色で塗る場合)
# PyMOL 2.3時代まで
util.color_deep("red", '1alk', 0)
# PyMOL 2.4以降(予定)
color_deep red, 1alk