大腸菌由来アルカリホスファターゼのグラフィック表示
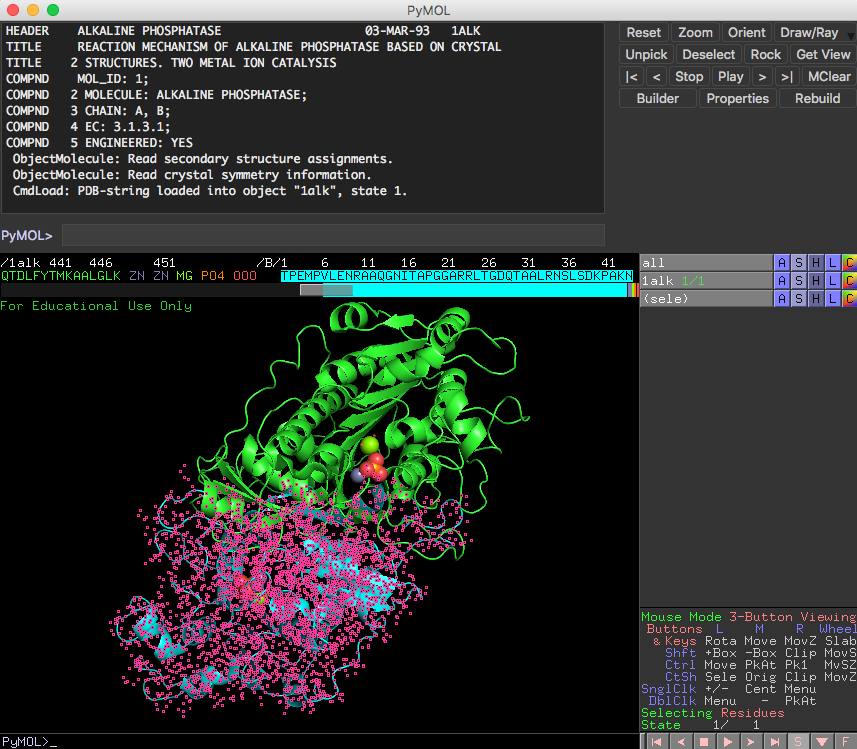
PyMOLを起動した後、画面左上にあるFileメニューからOpen...を選び、先程デスクトップに移動させた1alk.pdbファイルを選択します。すると、PyMOL画面の中にPDB ID: 1ALKの大腸菌由来アルカリホスファターゼが表示されます。

Important
上記のメニューによる選択以外にも、マウス操作で
1alk.pdbファイルをPyMOLのウィンドウにドラッグ&ドロップすることでも同様にPDBファイルを開くことができます。
このチュートリアルではPyMOLのGUI機能を駆使して、アルカリホスファターゼの2次構造の配置(フォールド)や、リン酸付近の原子・残基の配置などを調べてみます。必要に応じて第2章 PyMOLのGUIの使い方のマウス操作 とMouse Mode: 3-Button Viewing を読みます。マウスで分子をドラッグすることで、分子の回転、平行移動、拡大縮小などが行うことができます。
PyMOLでは、まずマウス(またはselectコマンド)で原子やアミノ酸を選択(select) し、続いて選択した範囲に対する 操作(Action) を指定します。選択された部分はピンクのマーカーで強調されるので、今何が「選択」されているかに常に注意します。また、操作のやり直しはできないので、操作を間違えた場合にはその都度それを上書きする形で表示をやり直します。

チェインごとの色分け
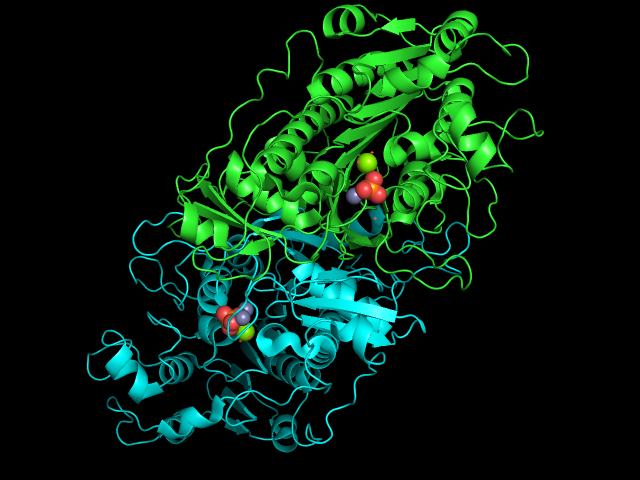
このアルカリホスファターゼがホモ2量体であることをわかりやすく表示するために、Aチェインを緑色(デフォルト)、Bチェインを水色(cyan)で表示してみます。PyMOL右下の画面にSEQという小さなボタンがあるので、ここを押すと、タンパク質が表示されている画面の上にアミノ酸配列が現れます(参考: 配列の表示について )。
今表示されたアミノ酸配列の上で、マウスを使ってチェインBをすべて選択します。次に、色の設定を参考にしてチェインBを水色に変更します(参考:色の設定 )。
動画バージョンです
※ PyMOLの画面上に存在する入力欄PyMOL>の所(中段上と一番下の2ヶ所に存在しますが、どちらに入力してもOK)で上や下の矢印キーを押すと、それまでに入力したコマンドを再び呼び出すことができます。また左右の矢印キーを使って、以前使ったコマンドを自由に編集できます。同じコマンドを何度も入力するのは面倒なので、上下左右の矢印キーを上手に使いましょう。
二次構造ごとの色分け
この大腸菌アルカリホスファターゼを、今度は二次構造による色分けを使って表示してみます(参考: 色の設定について )。
オブジェクトパネルにおける1ALKのCのところをクリックし、Colorのメニューのところで、[by ss]にマウスを重ねます。色分けの種類はデフォルトで3種類用意されており、どれを使っても問題ありませんが、ここでは一番上の例(ヘリックス:赤; シート:黄; ループ:緑)のカラーリングで表示してみます。
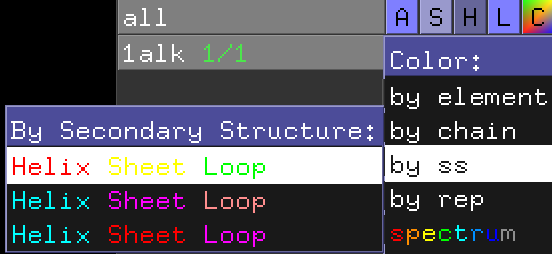
このように表示されるはずです。
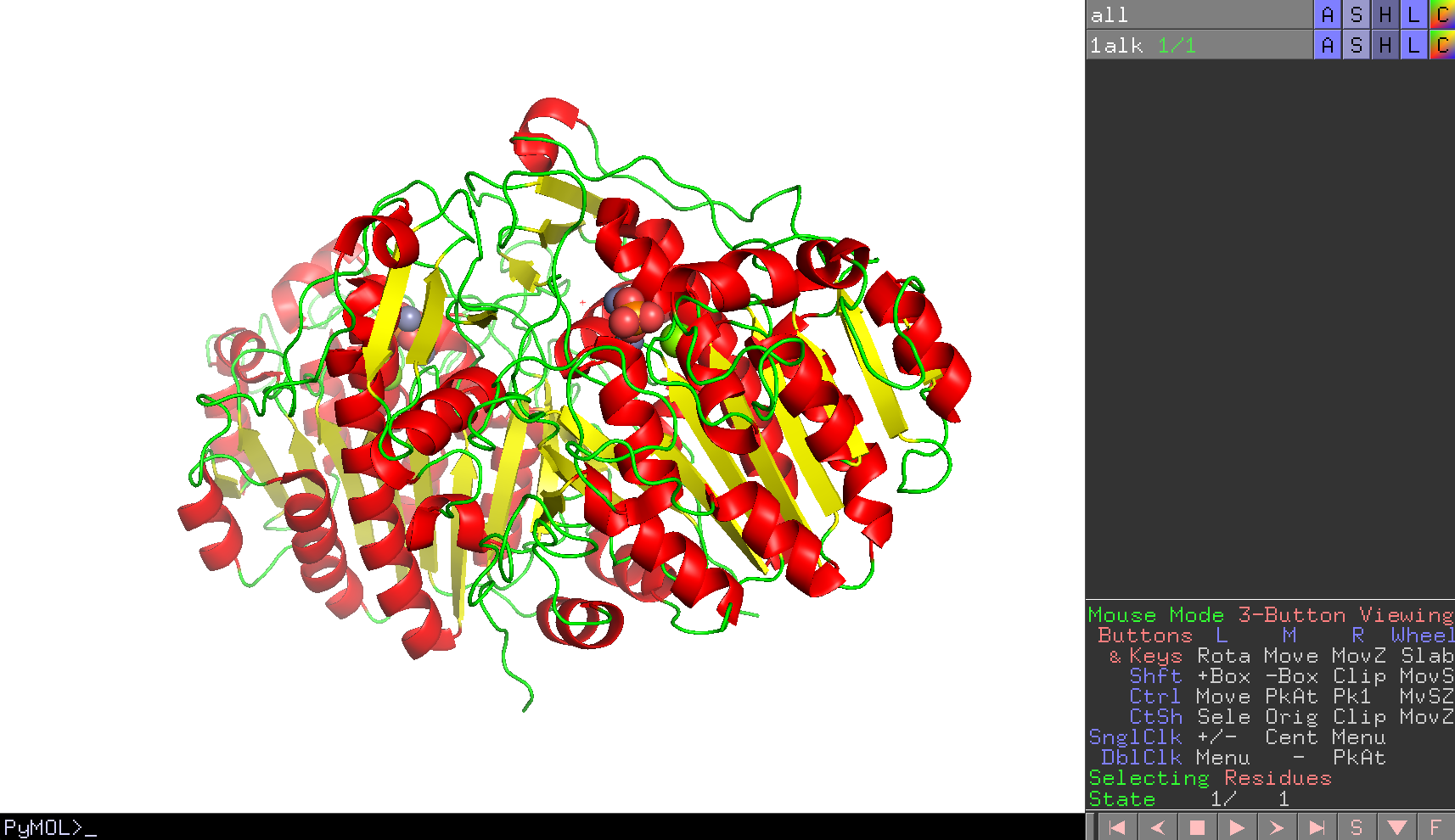
ここで、大腸菌アルカリホスファターゼの立体構造のCATHによる分類は、“Alpha Beta 3-Layer(aba) Sandwich“とされています。Cartoon表示で見る角度を調節して、このタンパク質の立体構造がCATHの分類通りにAlpha Betaの3層構造を形成していることを確認してみましょう。
画像の保存
画面に映されているタンパク質を画像ファイルとして保存します。ただし、そのまま保存しようとすると背景が黒のままとなってしまいます。
これを白背景で出力します。上部のDisplay > Backgroundメニューから、Whiteを選択します。
この設定の後、PyMOL画面の右上に存在するDraw/Rayボタンを押し、Draw (fast)またはRay (slow)ボタンを押すと、きれいな画像となって表示されます。
問題がなければ、Save Image to Fileボタンを押し、ファイル名を設定して保存しましょう。ちなみに、transparent backgroundにチェックを入れると背景透過画像を生成することができます(ただしRay (slow)を適用した場合のみ)。背景透過処理を行うとレポートやスライドの上で扱いやすくなるので、ぜひ利用しましょう。
その他の詳しい項目については2.7 画像の保存を参照してください。
セッションファイルへの保存
ここまでの作業内容をセッションファイルに保存します。セッションファイルとは、現在のPyMOLの画面の状態をそのまま保存しておくファイルのことです。画面上部にあるFileメニューからSave session as…を選んで名前を指定して保存します(kadai1.pseなど、わかりやすい名前をつけましょう)。(参考:セッションの保存 )。
保存したセッションファイルはFile > Openメニューから読み込むことができます。
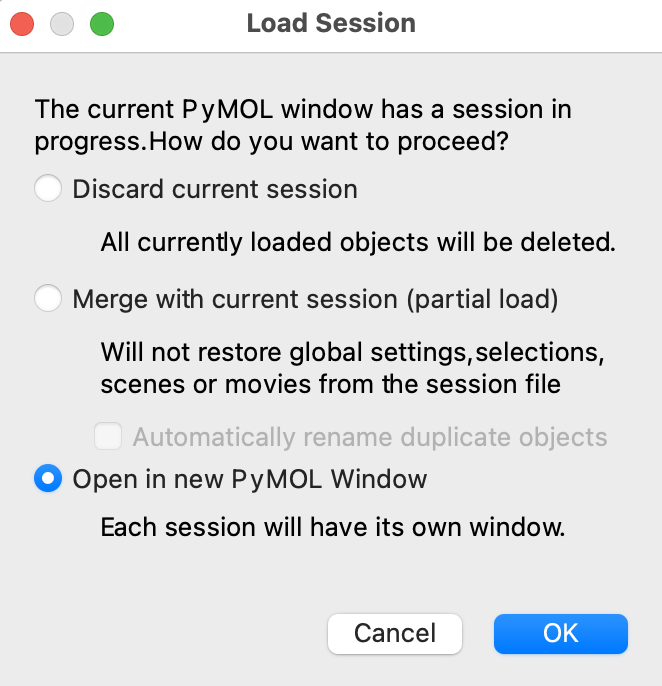
すでにタンパク質を他に表示させている状態でセッションファイルを読み込もうとすると下図のようなメッセージが現れますが、今開いている画面と別のPyMOL画面でタンパク質を表示させたい場合は、一番下のOpen in new PyMOL Windowを選択することをおすすめします。
※ (上級者向け)上述のPyMOL>の入力欄にコマンドを入力することでも可能です。デスクトップ上に保存したい場合は、cd ~/DesktopとしてWorking Directoryをデスクトップに設定した後、save kadai1.pseとすることでセッションファイルを保存できます。
リガンド結合部位への注目
ここから、タンパク質内のリン酸や金属が結合している部分に注目してみます。まず、全体構造の中のどこに結合しているかを確認してみます。
- PyMOL内で配列を表示させます。
- 表示された配列のところで、Chain Aの
ZN2つとMG,PO4をすべて選択します。 - 次に、オブジェクトパネルにおいてその選択範囲
(sele)についてStickとSpheresで表示させてみます(→分子構造の表示形式のON/OFF)。
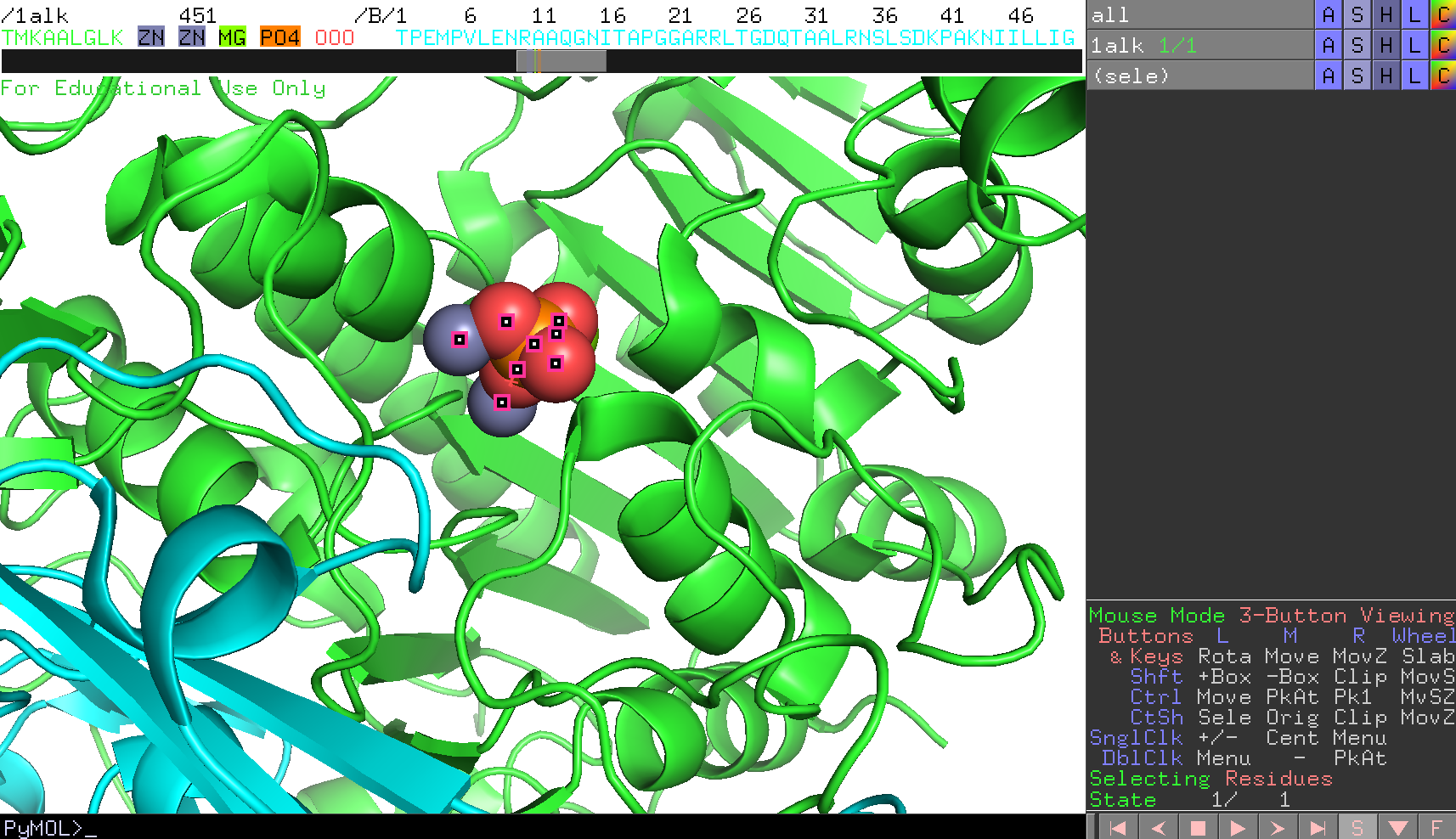
デフォルト設定では、sphere表示で表されるボールがかなり大きくなっているので、ここではいったん小さくしてみます。これはPyMOL >のところにコマンドを打つことで設定できます。
set sphere_scale, 0.4
これで球が小さく表示されました。
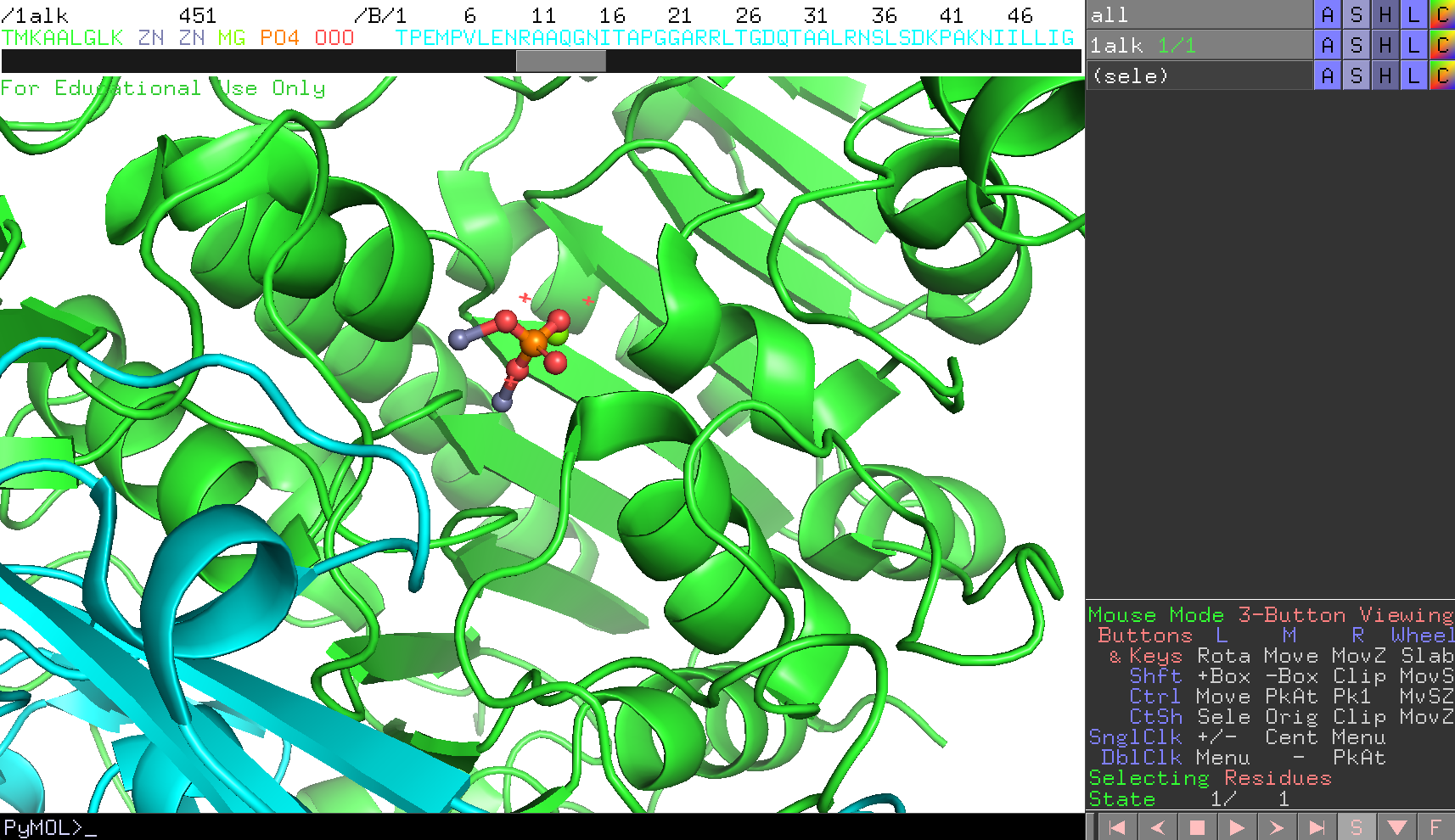
ここで、表示をチェインA(A鎖)だけにし、Bチェインはhideで隠します(→分子構造の表示形式のON/OFF)。リン酸や金属は各チェインに1組ずつ結合していることがわかります。
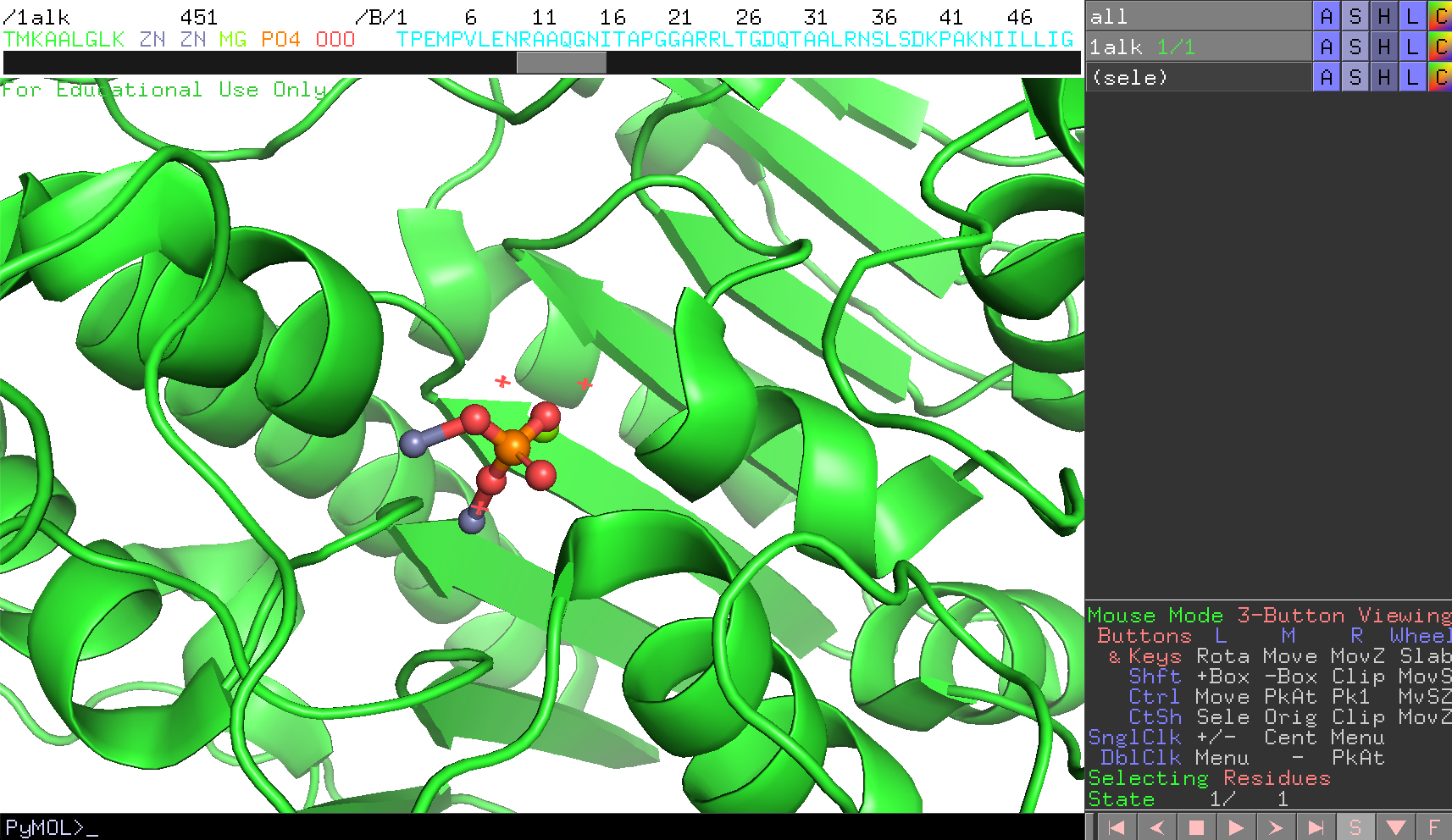
チェインAのリン酸(PO4)が画面の中心にくるよう設定してみます。これはこのPO4を選択した上で右クリックを押してメニューを表示させ、centerを選択することでできます。
参考:マウス操作と各モードにおける操作一覧
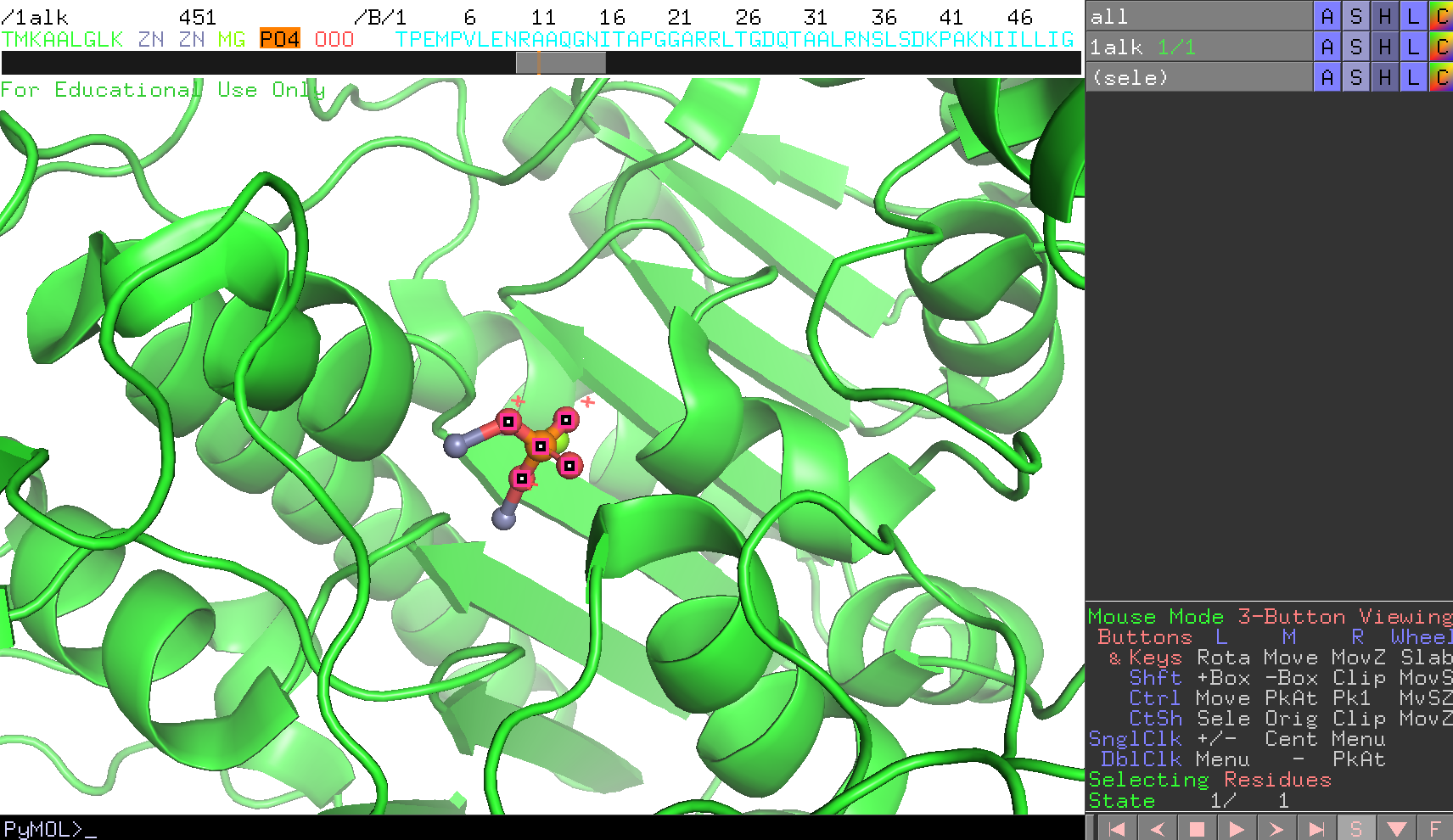
マウス操作で、画面を拡大して見やすくしておきます。
タンパク質のCartoon表示をオフにします(→分子構造の表示形式のON/OFF)。
チェインAのリン酸から4.6 Å以内にある水分子以外のアミノ酸残基を表示させます。チェインAのリン酸PO4だけが選択された状態で以下のコマンドを打ちます。
select byres resn * within 4.6 of sele
show sticks, sele
ここで、byresは「残基単位での選択」を意味する設定であり、とある残基が1原子でもリン酸から4.6 Åの距離以内に存在するのであれば、その残基をすべて選択するようにしています。その後、2行目のコマンドで選択した範囲をsticksで表示させます(オブジェクトパネルでも可能)
Zn,Mgは原子種ごとの色分け(→色の設定)にします。
リン酸付近にどのようなアミノ酸種が分布しているかを確認するために、先程の選択範囲(sele)について、ラベルの設定でlabel -> residuesを選択します。
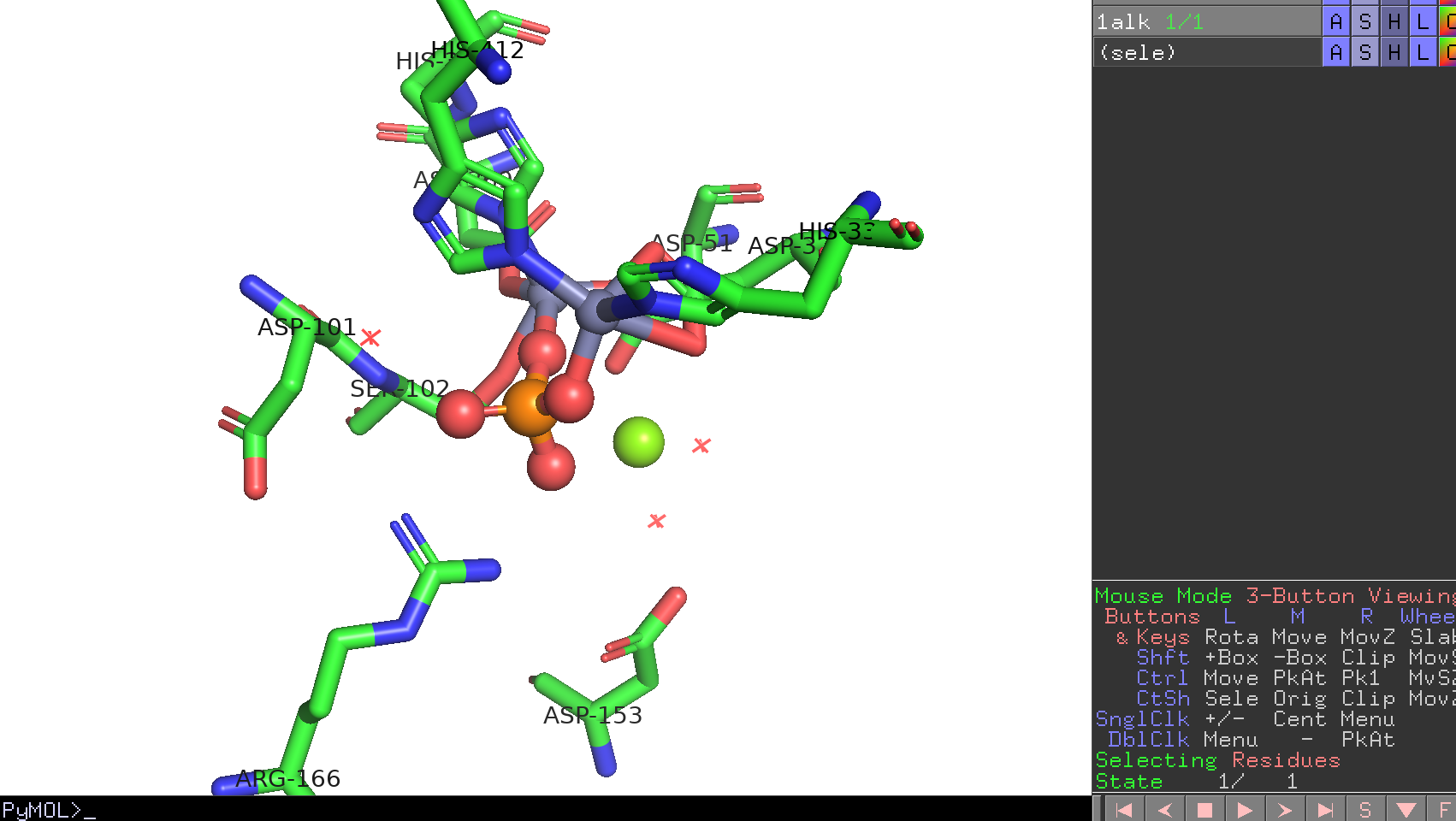
PyMOL画面を回転させて、どのアミノ酸がこのリガンド結合部位に存在しているかをすべてメモします。目視で行っても構いませんが、ここではPyMOLがPythonプログラミングで動いているということを利用して、以下のコマンドを入力して確認してみましょう。
コマンド入力欄に以下のコマンドを順次入れていきます(コピー&ペースト可能)。
# チェインAのリン酸を選択する(残基名がPO4でかつchain Aのものをselectする)
select resn PO4 and chain A
# 現在の選択範囲から4.6Å以内にある分子を、残基単位(byres)ですべて選択する
select byres resn * within 4.6 of sele
# ここで空の配列であるreslistを作成しておく(初期化)
reslist = []
# pymolのiterateコマンドを用いて、選択範囲(sele)に存在するCα炭素(name CA)について
# (residue_id, residue_name)のタプルをreslistに繰り返し加えていく
iterate sele and name CA, reslist.append((resi, resn))
# reslistを表示する
print(reslist)
ここまでうまく入力されていれば、最後に以下のように結果が表示されるはずです。
PyMOL> print(reslist)
[('51', 'ASP'), ('101', 'ASP'), ('102', 'SER'), ('153', 'ASP'), ('166', 'ARG'), ('327', 'ASP'), ('331', 'HIS'), ('369', 'ASP'), ('370', 'HIS'), ('412', 'HIS')]
これはpythonで言うところの、リスト型変数reslistの中にタプル型で(残基番号, 3文字残基名)の組が入っている形になっています。
これをよく生物学の表示で使われるようなMET-1, ASP-2のような表示に変換したい場合は、pythonのprint文を知識を使って例えば以下のようにpythonプログラムを書けばうまく出力することができます。ただし、PyMOLのコマンドラインでpythonプログラムを書く場合はpythonとpython endというブロックの間に挟む必要があります。(参考: https://pymolwiki.org/index.php/Python )
python
# リスト内のタプルをi, jに代入するfor loop
for i, j in reslist:
# print&format文法を使って"{residue_name}-{residue_id}"の順番で表示する
print("{0}-{1}".format(j, i))
python end
すると、以下のように表示されます。
PyMOL>python end
ASP-51
ASP-101
SER-102
ASP-153
ARG-166
ASP-327
HIS-331
ASP-369
HIS-370
HIS-412
こうすれば目視でやるより書き漏らしがなくて済みますね。
終わったら、再び現在までの作業内容をセッションファイルに保存しておきます。ファイル名はたとえば1alk_active_site.pseなどとしておくと分かりやすくて良いでしょう。