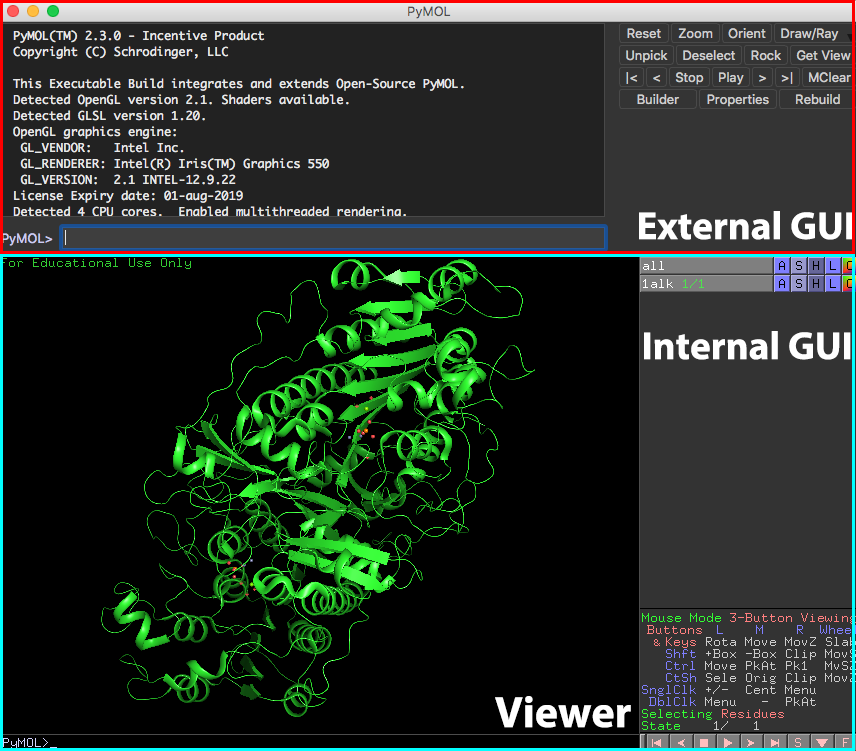
第2章 Internal GUIの使い方
GUIとはGraphical User Interfaceのことで、マウスを使ってタンパク質オブジェクトを操作したり、設定を変更したりできるユーザーインターフェースのことです。またPyMOLにはInternal GUIとExternal GUIと呼ばれるメニューが存在し、マウスのクリックを使った直感的な操作が行えるようになっています。
Press ← or → to navigate between chapters
Press S or / to search in the book
Press ? to show this help
Press Esc to hide this help
GUIとはGraphical User Interfaceのことで、マウスを使ってタンパク質オブジェクトを操作したり、設定を変更したりできるユーザーインターフェースのことです。またPyMOLにはInternal GUIとExternal GUIと呼ばれるメニューが存在し、マウスのクリックを使った直感的な操作が行えるようになっています。